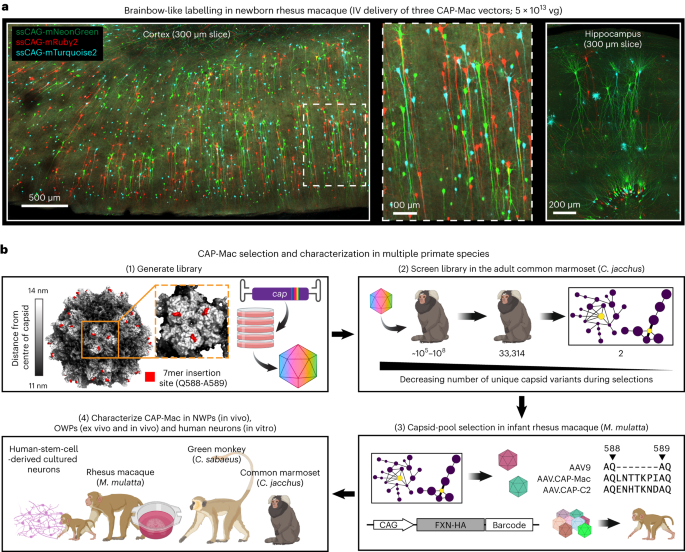
All the experiments and procedures were approved by local regulatory boards and committees and were required to comply with study protocols. All the mouse procedures were performed at Caltech, approved by the Caltech Institutional Animal Care and Use Committee (IACUC; protocol 1738). Marmoset (protocol TGC-03) and adult macaque (protocol LN-14) procedures took place at the NIH and were approved by the NIH IACUC. Marmoset procedures were also completed at University of California San Diego (UCSD) (protocol S09147) and were in compliance with and approved by the UCSD IACUC. Infant macaque procedures took place at the California National Primate Research Center at University of California Davis, and were approved by their local IACUC (protocol 22525). Green monkey procedures took place at Virscio and were approved by their local IACUC.
AAV DNA library generation
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jy89g2w/v1). We initially generated diversity at the DNA level, which we then used to produce transfection material to produce the AAV capsid library, as described previously in detail16. For the first-round library, we introduced this genetic diversity using primers containing degenerate nucleotides inserted between amino acids 588 and 589 (refs. 12,13,16) (VP1 numbering; Supplementary Fig. 1a). We used a reverse primer containing 7 degenerate nucleotides ([NNK] × 7) to randomly generate polymerase chain reaction (PCR) fragments containing unique 7mer sequences inserted into the cap gene. For the second-round DNA library, we used a synthetic oligo pool (Twist Bioscience) as a reverse primer, encoding only variants selected for further screening (total, 66,628 DNA oligos; 33,314 variants recovered after first-round selections plus a codon-modified replicate of each). All the reverse primers contained a 20 bp 5′ overhang complementary to the cap sequence near the AgeI restriction enzyme sequence and were paired with a forward primer containing a 20 bp 5′ overhang near the XbaI restriction enzyme sequence. We then inserted the PCR fragments containing the diversified region into the rAAV-ΔCAP-in-cis-Lox plasmid via Gibson assembly to generate the resulting AAV DNA library, namely, rAAV-CAP-in-cis-Lox, using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, E2621).
AAV capsid library production
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jyz9g2w/v1). We generated AAV capsid libraries according to previously published protocols16,70. Briefly, we transfected HEK293T cells (ATCC, cat # CRL-3216; RRID: CVCL_0063) in 150 mm tissue culture plates using transfection-grade linear polyethylenimine (PEI; Polysciences). In each plate, we transfected four plasmids: (1) the assembled rAAV-Cap-in-cis-Lox AAV DNA library, which is flanked by inverted terminal repeats required for AAV encapsidation; (2) AAV2/9 REP-AAP-ΔCAP, which encodes the REP and AAP supplemental proteins required for AAV production with the C terminus of the cap gene excised to prevent recombination with the AAV DNA library and subsequent production of replication-competent AAV; (3) pHelper, which encodes the necessary adenoviral proteins required for AAV production; and (4) pUC18 (Addgene ID: 50004; RRID: Addgene_50004), which contains no mammalian expression vector but is used as filler DNA to achieve the appropriate nitrogen-to-phosphate ratio for optimal PEI transfection. During preparation of the PEI–DNA mixture, we added 10 ng of our AAV DNA library (rAAV-Cap-in-cis-Lox) for every 150 mm dish and combined AAV2/9 REP-AAP-ΔCAP, pUC18 and pHelper in a 1:1:2 ratio (40 µg of total DNA per 150 mm dish). At 60 h post-transfection, we purified the AAV capsid library from both cell pellet and media using polyethylene glycol precipitation and iodixanol gradient ultracentrifugation. Using quantitative PCR, we then determined the titre of the AAV capsid libraries by amplifying DNaseI-resistant viral genomes relative to a linearized genome standard according to established protocols70.
Marmoset experiments
Capsid library selections
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). All the marmoset (C. jacchus) procedures were performed at the National Institute of Mental Health (NIMH) and approved by the local IACUC. Marmosets were born and raised in NIMH colonies and housed in family groups under standard conditions of 27 °C and 50% humidity. They were fed ad libitum and received enrichment as part of the primate enrichment program for NHPs at the NIH. For all the marmosets used in this study, there were no detectible neutralizing antibodies at a 1:5 serum dilution before IV infusions (assayed by the Penn Vector Core, University of Pennsylvania). They were then individually housed for several days and acclimated to a new room before injections. Four adult males were used for the library screening, two each for the first- and second-round libraries. The day before infusion, the animals’ food was removed. Animals were anaesthetized with isoflurane in oxygen, the skin over the femoral vein was shaved and sanitized with an isopropanol scrub and 2 × 1012 vg of the AAV capsid library was infused over several minutes. Anaesthesia was withdrawn and the animals were monitored until they became active, on which they were returned to their cages. Activity and behaviour were closely monitored over the next 3 days, with daily observations thereafter.
At 4 weeks post-injection, marmosets were euthanized (Euthanasia, VetOne) and perfused with 1× phosphate-buffered saline (PBS). After the first-round library, the brain was cut into four coronal blocks, flash frozen in 2-methylbutane (Sigma-Aldrich, M32631), chilled with dry ice and stored at −80 °C for long-term storage. After the second-round library, the brain was cut into six coronal blocks and, along with sections of the spinal cord and liver, was flash frozen and stored at −80 °C for long-term storage.
Individual characterization of AAVs in marmosets
The details of the procedures in this section can be found on protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 and https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Two adult common marmosets (C. jacchus) were used for this experiment: Conan (male, 2.8 years old, 0.386 kg) and Sandy (female, 5.8 years old, 0.468 kg) (Supplementary Table 3 provides more details). They were housed under standard conditions of 27 °C and 50% humidity, with ad libitum access to food and water. All the animals were group housed, and the experiments were performed in the Cortical Systems and Behavior Laboratory at UCSD. All the experiments were approved by the UCSD IACUC. The day before infusion, the animals’ food was removed.
Animals were anaesthetized with ketamine (Ketaset, Zoetis 043-304, 20 mg kg−1), the skin over the saphenous vein was shaved and sanitized with an isopropanol scrub and 2 × 1013 vg kg−1 of AAV was infused over 5 min. The animals were monitored until they became active, on which they were returned to their cages. Activity and behaviour were closely monitored over the next 3 days, with daily observations thereafter. Blood samples were taken on days 1, 7, 14, 21 and 31 to measure the viral concentration in plasma.
At 31 days post-injection, the marmosets were anaesthetized with ketamine as described earlier and then euthanized (Euthasol, Virbac 200-071, 1 ml kg−1) and perfused with 1× PBS. Brains and organs were cut in half, and one half was flash frozen in 2-methylbutane (Sigma-Aldrich, M32631), chilled with dry ice and stored at −80 °C. The other half was fixed in 4% paraformaldehyde (PFA) (Thermo Scientific, J19943-K2) overnight and then stored at 4 °C in PBS azide (Sigma-Aldrich, S2002-100G, 0.025%). The samples were then shipped to the California Institute of Technology (Caltech) for analysis. For GLUT1 staining, we incubated slices with rabbit anti-GLUT1 (1:200; Millipore-Sigma, cat # 07-1401; RRID: AB_1587074), performed three to five washes with PBS, incubated with donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) and washed three to five times before mounting. We diluted all antibodies and performed all incubations using PBS supplemented with 0.1% Triton X-100 (Sigma-Aldrich, T8787) and 10% normal donkey serum (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) overnight at room temperature with shaking.
Viral library DNA extraction and NGS sample preparation
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). We previously reported that viral library DNA and endogenous host RNA can be isolated using TRIzol by precipitating nucleic acid from the aqueous phase12,16. Therefore, to extract viral library DNA from marmoset tissue, we homogenized 100 mg of spinal cord, liver and each coronal block of the brain in TRIzol (Life Technologies, 15596) using a BeadBug (Benchmark Scientific, D1036) and isolated nucleic acids from the aqueous phase according to the manufacturer’s recommended protocol. We treated the reconstituted precipitate with RNase (Invitrogen, AM2288) and digested with SmaI to improve downstream viral DNA recovery via PCR. After digestion, we purified with a Zymo DNA Clean and Concentrator kit (D4033) according to the manufacturer’s recommended protocol and stored the purified viral DNA at −20 °C.
To append Illumina adaptors flanking the diversified region, we first PCR amplified the region containing our 7mer insertion using 50% of the total extracted viral DNA as a template (25 cycles). After Zymo DNA purification, we diluted the samples at 1:100 and further amplified around the variable region with ten cycles of PCR, appending binding regions for the next PCR reaction. Finally, we appended Illumina flow cell adaptors and unique indices using NEBNext Dual Index Primers (New England Biolabs, E7600) via ten more cycles of PCR. We then gel purified the final PCR products using a 2% low-melting-point agarose gel (Thermo Fisher Scientific, 16520050) and recovered the 210 bp band.
For the second-round library only, we also isolated the encapsidated AAV library ssDNA for NGS to calculate the library enrichment scores, a quantitative metric that we used to normalize for differences in titre of the various variants in our library (see ref. 16 and the ‘NGS read alignment, analysis and generation of network graphs’ section). To isolate the encapsidated viral genomes, we treated the AAV capsid library with DNaseI and digested capsids using proteinase K. We then purified the ssDNA using phenol–chloroform, amplified viral transgenes by two PCR amplification steps to add adaptors and indices for Illumina NGS and purified using gel electrophoresis. This viral library DNA, along with the viral DNA extracted from the tissue, was sent for deep sequencing using an Illumina HiSeq 2500 system (Millard and Muriel Jacobs Genetics and Genomics Laboratory, Caltech).
NGS read alignment, analysis and generation of network graphs
Raw FASTQ files from NGS runs were processed with custom-built scripts (https://github.com/GradinaruLab/protfarm and https://github.com/GradinaruLab/mCREATE)16. For the first-round library, the pipeline to process these datasets involved filtering to remove low-quality reads, utilizing a quality score for each sequence, and eliminating bias from PCR-induced mutations or high GC-content. The filtered dataset was then aligned by a perfect string match algorithm and trimmed to improve the alignment quality. We then displayed the absolute read counts for each variant during the sequencing run within each tissue, and all the 33,314 variants that were found in the brain were chosen for the second-round selections.
After the second-round selections, we performed the same analysis to display the variant absolute read count of the injected virus library and of each variant within each tissue. Additionally, we calculated the library enrichment16 for each variant within each tissue:
$${overline{{rm{RC}}}}_{x,{rm{injected}},{rm{library}}}=,frac{{{rm{RC}}}_{x,{rm{injected}},{rm{library}}}}{mathop{sum }nolimits_{i=1}^{{N}_{{rm{injected}},{rm{library}}}}{{rm{RC}}}_{i,{rm{injected},{library}}}},$$
(1)
$${overline{{rm{RC}}}}_{x,{rm{tissue}}}=,frac{{{rm{RC}}}_{x,{rm{virus}}}}{mathop{sum }nolimits_{i=1}^{{N}_{{rm{tissue}}}}{{rm{RC}}}_{i,{rm{tissue}}}},$$
(2)
$${rm{Library},{enrichment}}=,{log }_{10}left(frac{{overline{{rm{RC}}}}_{x,rm{{injected},{library}}}}{{overline{{rm{RC}}}}_{x,{rm{tissue}}}}right),$$
(3)
such that for a given sample y (for example, the injected virus library or a tissue sample), RCx,y is the absolute read count of variant x, Ny is the total number of variants recovered and ({overline{{rm{RC}}}}_{x,{y}}) is the normalized read count.
To construct the CAP-Mac sequence clustering graph, we filtered the second-round NGS data based on the following criteria: (1) ≥100 read count in the injected library sample (24,186/33,314 variants), (2) ≥0.7 library enrichment score in more than two brain samples (415 variants) and (3) at least two more brain samples with ≥0.7 library enrichment than brain samples with less than −0.7 library enrichment (323 variants). To construct the CAP-C2 sequence graph, we filtered the second-round NGS data based on the following criteria: (1) ≥100 read count in the injected library sample and (2) both codon replicates present in at least two brain samples with ≥0.7 library enrichment (95 variants). These variants were then independently processed to determine pair-wise reverse Hamming distances (https://github.com/GradinaruLab/mCREATE) and clustered using Cytoscape (v. 3.9.0; RRID: SCR_003032) as described previously in detail16. Networks presented show capsid variants (nodes) connected by edges if the pair-wise reverse Hamming distance is ≥3.
Cloning individual AAV capsid variants
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.n2bvj87ebgk5/v1). For single-variant characterization, we cloned new variant plasmids by digesting a modified version of the pUCmini-iCAP-PHP.eB (Addgene ID: 103005; RRID: Addgene_103005) backbone using MscI and AgeI. We designed a 100 bp primer that contained the desired 21 bp insertion for each capsid variant and the regions complementary to the AAV9 template with ~20 bp overlapping the digested backbone. We then assembled the variant plasmid using NEBuilder HiFi DNA Assembly Master Mix, combining 5 μl of 200 nM primer with 30 ng of digested backbone in the reaction mixture. The capsid plasmid used to produce AAV.CAP-Mac is available on Addgene (Addgene ID: 200658; RRID: Addgene_200658).
Individual AAV production and purification
The details of this procedure can be found on protocols.io (https://doi.org/10.17504/protocols.io.14egn2dqzg5d/v1). To produce variants for pool testing, we followed our previously published protocol70 using 150 mm tissue culture dishes. For individual AAV.CAP-Mac and AAV9 characterization in vivo and in vitro, we adopted our published protocol to utilize ten-layer CellSTACKs (Corning, 3320) to efficiently produce viruses at a high titre to dose rhesus macaques and green monkeys. Specifically, we passaged twenty 150 mm dishes at approximately 70% confluency into a ten-layer CellSTACK 24 h before transfection. On the day of the transfection, we prepared the PEI–DNA transfection mixture for forty 150 mm dishes and combined the transfection mixture with media and performed a complete media change for the CellSTACK. We collected and changed the media at 72 h post-transfection similar to production in 150 mm dishes. At 120 h post-transfection, we added ethylenediaminetetraacetic acid (Invitrogen, 15575020) to a final concentration of 10 mM and incubated at 37 °C for 20 min, occasionally swirling and tapping the sides of the CellSTACK to detach the cells. We then removed the media and cell mixture and proceeded with the AAV purification protocol70. Of note, during the buffer exchange step after ultracentifugation, we used centrifugal protein concentrators with polyethersulfone membranes (Thermo Scientific, 88533) instead of Amicon filtration devices and used Dulbecco’s PBS supplemented with 0.001% Pluronic F-68 (Gibco, 24040032).
Rodent experiments
All the rodent procedures were performed at Caltech and were approved by the local IACUC. We purchased C57BL/6J (strain #: 000664; RRID: IMSR_JAX:000664), BALB/cJ (strain #: 000651; RRID: IMSR_JAX:000651) and DBA/2J (strain #: 000671; RRID: IMSR_JAX:000671) mice (all males, 6–8 weeks old) from The Jackson Laboratory. For IV administration in mice, we delivered 5 × 1011 vg of virus through the retro-orbital sinus70,71 using a 31 gauge insulin syringe (BD, 328438). See protocols.io for more details on retro-orbital injections of AAV in mice (https://doi.org/10.17504/protocols.io.3byl4joy8lo5/v1). For intracerebroventricular (ICV) administration in mice, we injected 5.0 × 1010 or 1.5 × 1011 vg into the lateral ventricle. Briefly, we anaesthetized mice using isoflurane (5% for induction, 1–3% for maintenance) with 95% O2/5% CO2 (1 l min−1) and the mice were head fixed in a stereotaxic frame. After shaving the head and sterilizing the area with chlorohexidine, we subcutaneously administered 0.05 ml of 2.5 mg ml−1 bupivacaine, and a midline incision was made and the skull was cleaned of blood and connective tissue. After levelling the head, burr holes were bilaterally drilled above the lateral ventricles (0.60 mm posterior to bregma and 1.15 mm from the midline). Viral vectors were aspirated into 10 µl NanoFil syringes (World Precision Instruments) using a 33 gauge microinjection needle, and the needle was slowly lowered into the lateral ventricle (1.6 mm from the pial surface). The needle was allowed to sit in place for approximately 5 min and 3–5 µl of viral vector was injected using a microsyringe pump (World Precision Instruments, UMP3) and pump controller (World Precision Instruments, Mircro3) at a rate of 300 nl min−1. All the mice intraoperatively received 1 mg kg−1 of buprenorphine SR and 5 mg kg−1 of ketoprofen subcutaneously and 30 mg kg−1 of ibuprofen and 60 mg kg−1 of trimethoprim/sulfamethoxazole for 5 days post-surgery. See protocols.io for more details on ICV injections of AAV in mice (https://doi.org/10.17504/protocols.io.5qpvorm4dv4o/v1). After 3 weeks of expression, all the mice were perfused with PBS and fixed in 4% PFA. All the organs were extracted, incubated in 4.00% PFA overnight, transferred into PBS supplemented with 0.01% sodium azide and stored at 4 °C for long-term storage. We sliced the brain into 100 μm sections with a vibratome (Leica Biosystems, VT1200S), mounted in Prolong Diamond Antifade (Invitrogen, P36970) and imaged using a confocal microscope (Zeiss, LSM 880) using ZEN (Black edition). See protocols.io for more details on tissue handling (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 and https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1).
Rhesus macaque experiments
The details of the procedures in this section can be found on protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1). Neonate macaques (0.45–1.40 kg) were weaned at birth. Within the first month, macaques were infused with AAV vectors either intravenously or intrathecally. All adult macaques (8–17 years old; 4.65–11 kg) included in this study were infused with AAV via IV administration only. For IV injections, the animals were anaesthetized with ketamine (0.10 ml) and the skin over the saphenous vein was shaved and sanitized. AAV (between 2 × 1013 and 1 × 1014 vg kg−1) was slowly infused into the saphenous vein over ~1 min in <0.75 ml of PBS. For ICM injections, the animals were intramuscularly administered a sedative and the area of the skin at the neck was shaved and aseptically prepared. A needle was advanced into the cisterna magna to remove a small amount of CSF proportional to the amount of fluid injected. Then, a sterile syringe containing the sterile preparation of the AAV (1.5 × 1012 or 2.5 × 1013 vg kg−1) proportional to the amount of fluid collected was aseptically attached and slowly injected. All the animals were monitored during recovery from sedation throughout the day and then daily for any adverse findings. All the monkeys were individually housed within sight and sound of conspecifics. Tissue was collected 4–11 weeks after injection. The animals were deeply anaesthetized and received sodium pentobarbital in accordance with guidelines for humane euthanasia of animals at the California National Primate Research Center. All the material injected into rhesus macaques was free of endotoxins (<0.1 EU ml−1), and protein purity was confirmed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis. Supplementary Tables 4 and 5 list the route of administration, AAV variants, viral dose, genetic cargo and duration of expression for each experiment.
Pool testing in rhesus macaques
The details of the procedures in this section can be found on protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1, https://doi.org/10.17504/protocols.io.3byl4jo68lo5/v1 and https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Neonate macaque pool experiments (RM-001 to RM-004) were performed at the CNPRC at UC Davis and approved by the local IACUC. Adult macaque pool experiments (RMN-001 and RMN-002) were performed at the NIMH and approved by their local IACUC. Macaques were perfused with ice-cold RNase-free PBS. At the time of perfusion, one hemisphere of the brain was flash frozen and the other hemisphere was sectioned into 4 mm coronal blocks and post-fixed in 4% PFA for 48 h and transferred to Caltech for further processing. For HA staining, we incubated slices with rabbit anti-HA (1:200; Cell Signaling Technology, cat # 3724; RRID: AB_1549585), performed three to five washes with PBS, incubated with donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) and washed three to five times before mounting. We diluted all the antibodies and performed all the incubations using PBS supplemented with 0.1% Triton X-100 (Sigma-Aldrich, T8787) and 10% normal donkey serum (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) overnight at room temperature with shaking.
To isolate the viral DNA and whole RNA, 100 mg slices from the brain and liver were homogenized in TRIzol (Life Technologies, 15596) using a BeadBug (Benchmark Scientific, D1036) and the total DNA and RNA were recovered according to the manufacturer’s recommended protocol. The recovered DNA was treated with RNase, restriction digested with SmaI and purified with a Zymo DNA Clean and Concentrator kit (D4033). The recovered RNA was treated with DNase, and cDNA was generated from the mRNA using SuperScript III (Thermo Fisher Scientific, 18080093) and oligo(dT) primers according to the manufacturer’s recommended protocol. We used PCR to amplify the barcode region using 50 ng of viral DNA or cDNA as the template. After Zymo DNA purification, we diluted the samples at 1:100 and further amplified the barcode region using primers to append adaptors for Illumina NGS. After cleanup, these products were further amplified using NEBNext Dual Index Primers for Illumina sequencing (New England Biolabs, E7600) for ten cycles. We then gel purified the final PCR products using a 2% low-melting-point agarose gel (Thermo Fisher Scientific, 16520050). Pool testing enrichment was calculated identically to library enrichment, but is represented in Fig. 2b,c on a linear scale.
Individual characterization of CAP-Mac in rhesus macaques
The details of the procedures in this section can be found on protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 and https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Neonate macaques were perfused with PBS and 4% PFA. The brain was sectioned into 4 mm coronal blocks and all the tissue was post-fixed in 4% PFA for 3 days before storage in PBS. The single adult macaque used for in vivo, individual characterization (RM-020; 17 years old, 11 kg) was perfused with RNase-free PBS, and one half-hemisphere was flash-frozen and the other sectioned into 4 mm coronal blocks and post-fixed in 4% PFA. All the tissue was transferred to Caltech for further processing. Brains and livers were sectioned into 100 μm slices using a vibratome. Additionally, sections of brain and spinal cord were incubated in 30% sucrose overnight and embedded in O.C.T. compound (Scigen, 4586) and sectioned into 50 μm slices using a cryostat (Leica Biosystems, CM1950). All the slices were mounted using Prolong Diamond Antifade and imaged using a confocal microscope. For GFP staining of the spinal cord and brain slices from the intrathecally administered macaque, we incubated slices with chicken anti-GFP (1:500; Aves Labs, cat # GFP-1020; RRID: AB_10000240), performed three to five washes with PBS, incubated with donkey anti-chicken IgY (1:200; Jackson ImmunoResearch Lab, cat # 703-605-155; RRID: AB_2340379) and washed three to five times before mounting. We diluted all the antibodies and performed all the incubations using PBS supplemented with 0.1% Triton X-100 (Sigma-Aldrich, T8787) and 10% normal donkey serum (Jackson ImmunoResearch, 017-000-121) overnight at room temperature with shaking.
For morphological reconstruction, we sectioned brains into 300 μm sections and incubated them in a refractive index matching solution72 for 72 h before mounting on a slide immersed in the refractive index matching solution. We imaged using a confocal microscope and ×25 objective (LD LCI Plan-Apochromat ×25/0.8 Imm Corr DIC) using 100% glycerol as the immersion fluid. We captured tiled Z stacks (1,024 × 1,024 for each frame using the suggested capture settings) around cells of interest and cropped appropriate fields of view for tracing. Tracing was done in Imaris (Oxford Instruments; RRID: SCR_007370) using semi-automated and automated methods.
For neuron (NeuN) and astrocyte (S100β) quantification, the slices were stained using anti-NeuN (EPR12763) antibody (1:200; Abcam, cat # ab177487; RRID: AB_2532109) or anti-S100β antibody (1:200; Abcam, cat # ab52642; RRID: AB_882426) overnight in PBS supplemented with 0.1% Triton X-100 and 10% normal donkey serum. The slices were washed three to five times with PBS and incubated overnight in anti-rabbit IgG antibody conjugated with Alexa Fluor 647 (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) in PBS + 0.1% Triton X-100 + 10% normal donkey serum. After three to five washes and mounting using Prolong Diamond Antifade, we obtained Z stacks using a confocal microscope and a ×25 objective. We segmented NeuN- and XFP-positive cells using custom scripts in Python (RRID: SCR_008394) and Cellpose (https://www.cellpose.org/; RRID: SCR_021716)73.
Ex vivo two-photon imaging
Brain slices of sizes suitable for imaging were prepared with a thickness of 400 µm from larger slices using a vibratome and stored in artificial cerebrospinal fluid bubbled with carbogen gas before two-photon imaging, as previously described in published protocols74,75. For testing GCaMP8s responses, electrical stimulation (4–5 V, 80 Hz, 0.3 s duration) with the indicated number of pulses was delivered using an extracellular monopolar electrode placed 100–200 µm away from the neuron imaged. The frame rate of imaging was 30 Hz. Traces of segmented regions of interest were plotted as ΔF/F0 = (F(t) − F0)/F0, where F0 is defined as the average of all the fluorescence values before the electrical stimulation. The rise time was defined as the time required for the rising phase of the signal to reach from 10% of the peak to 90% of the peak. The decay time constant was obtained by fitting the sums of exponentials to the decay phase of the signal. The signal-to-noise ratio was obtained by dividing the peak amplitude of the signal by the standard deviation of the fluorescence trace before the electrical stimulation.
Characterization in adult rhesus macaque slice
One adult rhesus macaque (14 years and 1 month old; 10.83 kg) from the Washington National Primate Research Center was planned for routine euthanasia, and the brain was collected as part of the facility’s Tissue Distribution Program. A block of the superior temporal gyrus was sectioned into 300 μm slices and the slices were recovered74 and cultured on an air–liquid membrane interface, as previously described76. Approximately 30 min after plating the slices, we administered 1–2 μl of AAV (5 × 1013 vg ml−1 of AAV9 or AAV.CAP-Mac packaging either ssCAG-FXN-HA or ssCAG-eGFP). The experiments were performed in biological triplicates for each condition and the culture medium was refreshed every 48 h until tissue collection at 8 days post-transduction. On the day of tissue collection, the slices were imaged to confirm transduction, slices were cut in half and each half slice was flash frozen in a dry ice–ethanol bath. The samples were stored at −20 °C until further processing.
Each half slice was processed (one each for DNA and RNA recovery). DNA was isolated using the Qiagen DNeasy Blood and Tissue kit (Qiagen, catalogue # 69504) and RNA was recovered using TRIzol (Thermo Fisher Scientific, catalogue # 15596026) and the PureLink RNA Mini kit (Thermo Fisher Scientific, catalogue # 12183018A). DNA was removed from the RNA sample by modifying the first wash of the PureLink RNA Mini kit as follows: wash with 350 µl of Wash Buffer 1, then add 80 µl of RNase-free DNaseI in RDD buffer (Qiagen catalogue # 79254) and incubate the column at room temperature for 15 min; then, wash again with 350 µl of Wash Buffer 1 before proceeding with the protocol. We performed first-strand cDNA synthesis from 400 ng total RNA in 20 µl reactions using a Promega GoScript Reverse Transcription kit (Promega, catalogue # A5000).
We then evaluated the vector genomes and viral transcripts found in each sample using quantitative PCR on a Roche Lightcycler II. Here 100 ng of DNA was used in a 20 µl amplification reaction using TaqMan probes from Thermo Fisher Scientific (EGFP-FAM probe, assay ID Mr04097229_mr, catalogue #4331182; custom genomic reference probe CN2386-2-VIC, Assay ID ARH6DUK, catalogue #4448512, designed to target both M. mulatta and Macaca nemestrina).
Green monkey experiments
All the green monkey (C. sabaeus) procedures were performed at Virscio and approved by their IACUC. All the monkeys were screened for neutralizing antibodies and confirmed to have <1:5 titre. At approximately 7–8 months of age (1.0–1.3 kg), the monkeys were intravenously dosed (Supplementary Table 6). Dose formulations were allowed to equilibrate to approximately room temperature for at least 10 min, but no more than 60 min before dosing. The IV dose volumes were based on day 0 body weights. The animals were sedated with ketamine (8.0 mg kg−1) and xylazine (1.6 mg kg−1). The injection area was shaved and prepped with chlorohexidine and 70% isopropanol and surgically scrubbed before insertion of the IV catheter. Dosing occurred with a single IV infusion of AAV (7.5 × 1013 or 7.6 × 1013 vg kg−1) on day 0 via the saphenous vein administered using a hand-held infusion device at a target rate of 1 ml min−1. General well being was confirmed twice daily by cage-side observation beginning 1 week before dosing. At the scheduled euthanisia time, the monkeys were sedated with ketamine (8–10 mg kg−1 intramuscular) and euthanized with sodium pentobarbital (100 mg kg−1 IV to effect). On loss of corneal reflex, a transcardiac perfusion (left ventricle) was performed with chilled PBS using a peristaltic pump set at a rate of approximately 100 ml min−1 until the escaping fluid ran clear before tissue collection. Cubes of tissue were collected from the left brain hemisphere and various other organs and frozen in the vapour phase of liquid nitrogen for further processing for biodistribution. The right brain hemisphere was removed and cut into ~4 mm coronal slices and post-fixed intact with approximately 20 volumes of 10% neutral-buffered formalin for approximately 24 h at room temperature.
Genomic DNA was extracted from CNS and peripheral tissues using the Thermo Fisher MagMax DNA Ultra 2.0 extraction kit (catalogue # A36570). DNA was assessed for yield by fluorometric quantification with the Qubit dsDNA assay. Approximately 20 ng of DNA was loaded into each 20 μl reaction and the plates were run on the BioRad CFX Connect Real-Time PCR Detection System (catalogue # 1855201). The viral copy number assay was validated for specificity by the detection of a single amplified product; sensitivity, by assessing the lower limit of detection to be greater than ten copies per reaction; and linearity, by ensuring the standard curve R2 was >0.95. Reactions were assembled in FastStart Universal SYBR Green Master (Rox) (catalogue catalogue # 4913850001). The sequences of the primers were ACGACTTCTTCAAGTCCGCC (forward) and TCTTGTAGTTGCCGTCGTCC (reverse). The PCR protocol used an initial denaturation step of 95 °C for 180 s, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s, with an imaging step following each 60 °C cycle. A standard curve was generated with linearized plasmid containing the GFP template sequence present in the virus from 1 × 108 to 1 × 1010 copies, diluted in naïve untreated macaque DNA samples prepared using an identical kit as the samples in this study to control for matrix effects. Copies of viral DNA were calculated from the standard curve using the equation for the line of the best fit. Multiplicity of infection values were calculated based on the measured total genomic weight of the host cell DNA per reaction.
Post fixation, the tissues were placed into 10% > 20% > 30% sucrose for 24 h each at 4 °C and then embedded in O.C.T. compound and stored at −80 °C until cryosectioning. The tissue blocks were brought up to −20 °C in a cryostat before sectioning into 30 μm slices and dry mounted onto slides after cryosectioning. After sectioning, the slides were left at room temperature overnight to dry. To assist in neuron quantification, we stained sections with the following antibodies and concentrations: rabbit anti-GFP (1:100; Millipore-Sigma, cat # AB3080; RRID: AB_91337) and mouse anti-NeuN (A60) (1:500; Millipore-Sigma, cat # MAB377; RRID: AB_2298772). For secondary antibody staining, the following secondary antibodies and concentrations were used: donkey anti-rabbit Alexa Fluor 488 (1:500; Thermo Fisher Scientific, cat # A-21206; RRID: AB_2535792) and donkey anti-mouse Alexa Fluor 647 (1:500; Thermo Fisher Scientific, cat # A-31571; RRID: AB_162542). All the antibodies were diluted with 1× PBS supplemented with 0.25% Triton X-100 (PBST) and 5.00% normal donkey serum. Primary antibody incubations were left overnight at room temperature. Sections were then washed with PBST. Secondary antibody incubations were performed for 2 h at room temperature. The sections were washed thrice in PBST. The sections were incubated in DAPI solution (1:10,000; Invitrogen, D1306) at room temperature for 5 min and then washed. Sections were coverslipped using Prolong Diamond Antifade.
Three sections per animal were stained and imaged. Each section was imaged in triplicate, with each region of interest having a total of nine images. Tissue regions of interest were imaged with a Keyence BZ-X800 with the following acquisition parameters: GFP (1/500 s), Cy5 (1 s), DAPI (1/12 s), high-resolution Z stack at 1.2 µm pitch. The following brain subregions were imaged: frontal, parietal, temporal, occipital cortices, cerebellum, caudate, putamen and thalamus (medial, ventral lateral and ventral posterior nuclei). A semi-automated cell-counting method was performed using ImageJ (RRID: SCR_003070) for quantification. Using thresholds and particle analysis, we quantified NeuN-positive and DAPI-positive cells. Using ImageJ’s cell counter, we manually counted GFP-positive cells as well as GFP and NeuN double-positive cells.
iPSC experiments
Neuronal cultures were produced by differentiating and maturing iPSC-derived neural progenitor cells with Stemdiff Forebrain Differentiation and Maturation kits (StemCell # 08600 and # 08605, respectively), according to their manufacturer’s protocols. Neural progenitor cells were produced by differentiation of the foreskin fibroblast-derived iPSC line: ACS-1019 (ATCC# DYS-0100; RRID: CVCL_X499), with Stemdiff SMADi Neural Induction kits (StemCell l#08581), selection with Stemdiff Neural Rosette Selection Reagent (StemCell l#05832) and expansion in Stemdiff Neural Progenitor Media (StemCell l#05833), according to their manufacturer’s protocols. Neurons were matured a minimum of 8 days before replating for transduction.
Mature neuronal cultures, seeded 15,000 cells per well in polyornithine- and laminin-coated black-walled 96-well optical plates, were cultured for an additional 4 days before transduction. Replicate wells were transduced with virus serially diluted across six orders of magnitude in 90% maturation media and 10% OptiPRO SFM. Four days post-transduction, the cultures were fixed with 4% PFA and counterstained with 1 µg ml−1 Hoechst 33322. The identification of transduced cells was determined by imaging 60 fields per well, using two-channel fluorescence detection (Hoechst, ex386/em440; eGFP, ex485/em521) on a CellInsight CX5 HCS Platform. Individual cells were identified by Hoechst detection of their nuclei and applying size- and contact-constrained ring masks to each cell. Cell transduction was determined by measuring the eGFP fluorescence above a threshold level within an individual ring mask. For each population, the percentage of transduced cells was plotted versus the applied dose. Curve fits and EC50 values were determined with Prism GraphPad (RRID: SCR_002798) agonist versus response (three-parameter) regression method. To report per-cell eGFP expression efficiencies, the eGFP spot fluorescence intensities were averaged from each ring mask across a minimum of 5,000 cells per well. The curve fits were obtained using the Prism GraphPad Biphasic X as the concentration regression method.
Statistics and reproducibility
For the representative images, at least three separate slices from each sample were mounted for imaging. Within each brain region of a single animal, at least three different fields of view were taken (minimum field of view after tiling, 2.38 mm × 2.38 mm; slice thickness, 50 µm), equating to nine separate fields of view across three brain slices, to ensure consistency across imaging samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Automotive / EVs, Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- BlockOffsets. Modernizing Environmental Offset Ownership. Access Here.
- Source: https://www.nature.com/articles/s41565-023-01419-x
- :is
- :where
- $UP
- 000
- 1
- 10
- 100
- 11
- 12
- 13
- 14
- 15%
- 16
- 17
- 180
- 2%
- 20
- 200
- 2011
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 24
- 25
- 27
- 30
- 300
- 31
- 33
- 39
- 40
- 50
- 500
- 60
- 66
- 7
- 70
- 72
- 75
- 8
- 80
- 9
- 95%
- a
- above
- Absolute
- access
- accordance
- According
- Achieve
- acquisition
- across
- active
- activity
- Ad
- add
- added
- Additional
- Additionally
- administered
- administration
- adopted
- Adult
- advanced
- adverse
- After
- again
- age
- AL
- Alexa
- algorithm
- aligned
- All
- allowed
- along
- also
- amount
- Amplification
- Amplified
- amplifying
- an
- analysis
- Anchor
- and
- animal
- animals
- Antibodies
- any
- applied
- Applying
- appropriate
- approved
- approximately
- AREA
- around
- article
- artificial
- AS
- assembled
- Assembly
- assessed
- Assessing
- assist
- At
- Automated
- available
- average
- away
- Backbone
- BAND
- based
- BD
- BE
- became
- before
- Beginning
- being
- Benchmark
- BEST
- between
- bias
- binding
- Black
- Block
- Blocks
- blood
- body
- born
- both
- BP
- Brain
- brains
- briefly
- brought
- buffer
- but
- by
- cages
- calculate
- calculated
- california
- CAN
- capture
- captured
- care
- Cargo
- CAT
- cell
- Cells
- cellular
- Center
- central
- chain
- chan
- change
- changed
- chosen
- clear
- click
- closely
- clustering
- collection
- Column
- combined
- combining
- committee
- Common
- complementary
- complete
- Completed
- compliance
- comply
- Compound
- concentration
- condition
- conditions
- Confirm
- CONFIRMED
- Connect
- connected
- constant
- construct
- contained
- contains
- control
- controller
- copies
- Core
- coronal
- Counter
- criteria
- Culture
- curve
- custom
- Custom-built
- Cut
- cycle
- cycles
- daily
- data
- datasets
- Davis
- day
- Days
- deep
- defined
- delivered
- delivery
- described
- Design
- designed
- desired
- details
- Detection
- Determine
- determined
- deviation
- device
- Devices
- Diamond
- Diego
- differences
- different
- dilution
- Display
- displayed
- distance
- distinct
- distribution
- diversified
- Diversity
- dna
- done
- dose
- dosing
- drive
- dry
- duration
- during
- e
- E&T
- each
- Earlier
- edition
- effect
- effects
- efficiencies
- efficient
- efficiently
- either
- elements
- eliminating
- embedded
- England
- ensure
- ensuring
- established
- Ether (ETH)
- EU
- evaluated
- Every
- example
- exchange
- expansion
- experiment
- experiments
- expression
- extract
- extraction
- family
- Fed
- female
- field
- Fields
- Fig
- Figure
- Files
- filtering
- final
- Finally
- findings
- First
- fit
- fitting
- five
- fixed
- Flash
- flow
- fluid
- followed
- following
- follows
- food
- For
- for yield
- Forward
- found
- four
- FRAME
- Free
- from
- frozen
- functional
- further
- GAS
- gauge
- General
- generate
- generated
- generation
- Genetics
- genome
- genomics
- given
- graph
- greater
- Green
- Group
- Group’s
- guidelines
- Half
- Handling
- Have
- having
- head
- Health
- here
- High
- high-resolution
- Holes
- homogenized
- host
- HTTPS
- human
- ICE
- ID
- identical
- Identification
- identified
- if
- ii
- iii
- images
- Imaging
- immersed
- immersion
- improve
- in
- included
- incubated
- independently
- index
- indicated
- Indices
- individual
- Individually
- induction
- infection
- information
- infused
- infusion
- initial
- initially
- instead
- Institute
- Institutional
- instruments
- interest
- Interface
- into
- intravenous
- introduced
- involved
- isolated
- Jackson
- kit
- lab
- laboratory
- Labs
- larger
- ld
- least
- left
- less
- Level
- libraries
- Library
- Life
- LIMIT
- Line
- LINK
- linked
- Liquid
- List
- Liver
- local
- long-term
- loss
- lower
- lowered
- made
- maintenance
- manually
- mapping
- mask
- Masks
- master
- Match
- matching
- material
- Matrix
- measure
- measured
- measuring
- Media
- medium
- mental
- Mental health
- method
- methods
- metric
- mice
- Microscope
- min
- minimum
- minutes
- mix
- mixture
- ML
- modified
- monitored
- Month
- months
- more
- mRNA
- MSCI
- namely
- nanotechnology
- National
- Nature
- Near
- necessary
- network
- networks
- Neural
- neuronal
- Neurons
- New
- next
- NIH
- no
- nodes
- normal
- number
- objective
- obtained
- occurred
- of
- Old
- on
- ONE
- only
- optimal
- optimized
- or
- orders
- Other
- our
- over
- overnight
- Oxford
- Oxygen
- packaging
- paired
- parameters
- part
- particle
- PBS
- PCR
- Peak
- Penn
- Pennsylvania
- per
- percentage
- perfect
- performed
- peripheral
- phase
- pipeline
- Pitch
- Place
- planned
- Plasma
- platform
- plato
- Plato Data Intelligence
- PlatoData
- plus
- pool
- population
- portfolio
- Precision
- preparation
- prepared
- present
- presented
- prevent
- previously
- primary
- primer
- probe
- procedure
- procedures
- process
- processed
- processing
- produce
- Produced
- Product
- Production
- Products
- progenitor
- Program
- Protective
- Protein
- Proteins
- protocol
- protocols
- provides
- published
- pump
- purchased
- Python
- quality
- quantitative
- Qubit
- Rabbit
- raised
- Rate
- ratio
- reach
- reaction
- reactions
- Read
- real-time
- received
- recommended
- recovery
- region
- regions
- regression
- regulatory
- relative
- remove
- Removed
- replicates
- report
- Reported
- Reporting
- representative
- represented
- required
- research
- respectively
- response
- responses
- restriction
- resulting
- reverse
- right
- Ring
- Rise
- rising
- RNA
- robust
- roche
- Room
- Route
- Run
- runs
- s
- same
- San
- San Diego
- sandy
- Scale
- scheduled
- SCI
- scientific
- score
- scores
- screening
- scripts
- secondary
- Section
- sections
- see
- segmentation
- selected
- selection
- selective
- Sensitivity
- sent
- separate
- Sequence
- sequencing
- Serum
- set
- settings
- several
- shipped
- show
- Sides
- Sight
- Signal
- similar
- single
- sit
- SIX
- sizes
- Skin
- Slice
- Slide
- Slides
- Slowly
- small
- sodium
- solution
- Sound
- specifically
- specificity
- Spot
- stack
- Stacks
- standard
- Step
- Steps
- storage
- stored
- String
- Study
- subsequent
- suitable
- superior
- Surface
- synthetic
- system
- systemic
- Systems
- table
- taken
- tapping
- Target
- targeted
- targeting
- Technologies
- Technology
- template
- ten
- Terminal
- Testing
- than
- that
- The
- The Area
- their
- Them
- then
- There.
- therefore
- These
- they
- this
- three
- threshold
- Through
- throughout
- time
- times
- tissues
- to
- took
- Total
- trace
- Tracing
- transfer
- transferred
- treated
- Triton
- Twice
- twist
- two
- types
- Ultra
- under
- unique
- Universal
- university
- University of California
- University of Pennsylvania
- until
- use
- used
- using
- utilize
- Utilizing
- validated
- Values
- Variant
- various
- version
- Versus
- via
- View
- viral
- virus
- viruses
- vivo
- volumes
- was
- washington
- Water
- we
- week
- Weeks
- weight
- WELL
- Wells
- were
- which
- whole
- widespread
- with
- within
- world
- X
- years
- Yield
- yields
- Zen
- zephyrnet