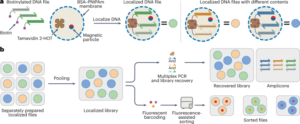
Toutes les expériences et procédures ont été approuvées par les conseils et comités de réglementation locaux et devaient se conformer aux protocoles d'étude. Toutes les procédures de souris ont été effectuées à Caltech, approuvées par le Caltech Institutional Animal Care and Use Committee (IACUC; protocole 1738). Les procédures de marmoset (protocole TGC-03) et de macaque adulte (protocole LN-14) ont eu lieu au NIH et ont été approuvées par le NIH IACUC. Les procédures de marmoset ont également été réalisées à l'Université de Californie à San Diego (UCSD) (protocole S09147) et étaient conformes et approuvées par l'UCSD IACUC. Les procédures pour les macaques infantiles ont eu lieu au California National Primate Research Center de l'Université de Californie à Davis et ont été approuvées par leur IACUC local (protocole 22525). Les procédures de singe vert ont eu lieu à Virscio et ont été approuvées par leur IACUC local.
Génération de bibliothèque d'ADN AAV
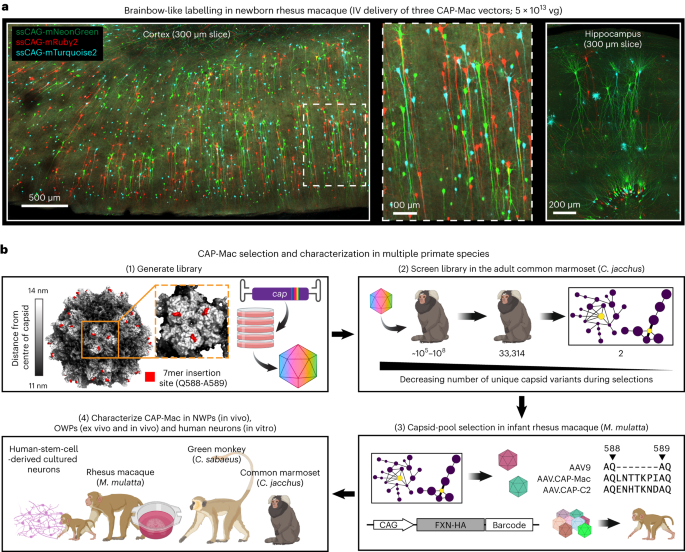
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jy89g2w/v1). Nous avons initialement généré une diversité au niveau de l'ADN, que nous avons ensuite utilisée pour produire du matériel de transfection afin de produire la bibliothèque de capsides AAV, comme décrit précédemment en détail.16. Pour la bibliothèque de premier tour, nous avons introduit cette diversité génétique en utilisant des amorces contenant des nucléotides dégénérés insérés entre les acides aminés 588 et 589 (réfs. 12,13,16) (numérotation VP1 ; Fig. 1a). Nous avons utilisé une amorce inverse contenant 7 nucléotides dégénérés ([NNK] × 7) pour générer de manière aléatoire des fragments de réaction en chaîne par polymérase (PCR) contenant des séquences 7mères uniques insérées dans le casquette gène. Pour la bibliothèque d'ADN de second tour, nous avons utilisé un pool d'oligos synthétiques (Twist Bioscience) comme amorce inverse, codant uniquement les variants sélectionnés pour un criblage ultérieur (total, 66,628 33,314 oligos d'ADN ; 20 5 variants récupérés après les sélections du premier tour plus un codon modifié réplique de chacun). Toutes les amorces inverses contenaient un surplomb XNUMX′ de XNUMX pb complémentaire du casquette séquence près de la séquence de l'enzyme de restriction AgeI et ont été appariés avec une amorce directe contenant un surplomb en 20 'de 5 pb près de la séquence de l'enzyme de restriction XbaI. Nous avons ensuite inséré les fragments PCR contenant la région diversifiée dans le plasmide rAAV-ΔCAP-in-cis-Lox via l'assemblage Gibson pour générer la bibliothèque d'ADN AAV résultante, à savoir rAAV-CAP-in-cis-Lox, à l'aide de NEBuilder HiFi DNA Assembly. Mélange maître (New England Biolabs, E2621).
Production de bibliothèques de capsides d'AAV
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jyz9g2w/v1). Nous avons généré des bibliothèques de capsides AAV selon des protocoles précédemment publiés16,70. En bref, nous avons transfecté des cellules HEK293T (ATCC, cat # CRL-3216; RRID: CVCL_0063) dans des plaques de culture tissulaire de 150 mm à l'aide de polyéthylèneimine linéaire de qualité transfection (PEI; Polysciences). Dans chaque plaque, nous avons transfecté quatre plasmides : (1) la bibliothèque d'ADN AAV rAAV-Cap-in-cis-Lox assemblée, qui est flanquée de répétitions terminales inversées nécessaires à l'encapsidation de l'AAV ; (2) AAV2/9 REP-AAP-ΔCAP, qui code les protéines supplémentaires REP et AAP nécessaires à la production d'AAV avec l'extrémité C-terminale du casquette gène excisé pour empêcher la recombinaison avec la banque d'ADN d'AAV et la production subséquente d'AAV compétent pour la réplication ; (3) pHelper, qui code les protéines adénovirales nécessaires à la production d'AAV ; et (4) pUC18 (Addgene ID : 50004 ; RRID : Addgene_50004), qui ne contient pas de vecteur d'expression de mammifère mais est utilisé comme ADN de remplissage pour obtenir le rapport azote/phosphate approprié pour une transfection PEI optimale. Lors de la préparation du mélange PEI-ADN, nous avons ajouté 10 ng de notre bibliothèque d'ADN AAV (rAAV-Cap-in-cis-Lox) pour chaque boîte de 150 mm et combiné AAV2/9 REP-AAP-ΔCAP, pUC18 et pHelper dans un Rapport 1:1:2 (40 µg d'ADN total par boîte de 150 mm). 60 h après la transfection, nous avons purifié la bibliothèque de capsides AAV à partir du culot cellulaire et du milieu en utilisant une précipitation au polyéthylène glycol et une ultracentrifugation en gradient d'iodixanol. Par PCR quantitative, nous avons ensuite déterminé le titre des bibliothèques de capsides d'AAV en amplifiant les génomes viraux résistants à la DNaseI par rapport à un standard de génome linéarisé selon des protocoles établis.70.
Expériences sur le ouistiti
Sélections de la bibliothèque de capsides
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Tous les marmousets (C. jacchus) les procédures ont été effectuées à l'Institut national de la santé mentale (NIMH) et approuvées par l'IACUC local. Les marmousets sont nés et ont grandi dans des colonies NIMH et logés en groupes familiaux dans des conditions standard de 27 ° C et 50% d'humidité. Ils ont été nourris à volonté et ont reçu un enrichissement dans le cadre du programme d'enrichissement des primates pour les PSN au NIH. Pour tous les ouistitis utilisés dans cette étude, il n'y avait pas d'anticorps neutralisants détectables à une dilution de sérum de 1:5 avant les perfusions IV (dosé par le Penn Vector Core, Université de Pennsylvanie). Ils ont ensuite été hébergés individuellement pendant plusieurs jours et acclimatés à une nouvelle pièce avant les injections. Quatre hommes adultes ont été utilisés pour le dépistage de la bibliothèque, deux pour les bibliothèques de premier et de second tour. La veille de l'infusion, la nourriture des animaux a été retirée. Les animaux ont été anesthésiés avec de l'isoflurane dans de l'oxygène, la peau au-dessus de la veine fémorale a été rasée et désinfectée avec un gommage à l'isopropanol et 2 × 1012 vg de la banque de capsides d'AAV a été infusé pendant plusieurs minutes. L'anesthésie a été retirée et les animaux ont été surveillés jusqu'à ce qu'ils deviennent actifs, après quoi ils ont été remis dans leurs cages. L'activité et le comportement ont été étroitement surveillés au cours des 3 jours suivants, avec des observations quotidiennes par la suite.
Quatre semaines après l'injection, les marmousets ont été euthanasiés (Euthanasia, VetOne) et perfusés avec une solution saline tamponnée au phosphate (PBS) 4 ×. Après la bibliothèque de premier tour, le cerveau a été découpé en quatre blocs coronaux, congelé dans du 1-méthylbutane (Sigma-Aldrich, M2), refroidi avec de la neige carbonique et stocké à -32631 ° C pour un stockage à long terme. Après la bibliothèque de deuxième tour, le cerveau a été découpé en six blocs coronaux et, avec des sections de la moelle épinière et du foie, a été congelé instantanément et stocké à -80 ° C pour un stockage à long terme.
Caractérisation individuelle des AAV chez les marmousets
Les détails des procédures de cette section peuvent être trouvés sur protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 et de https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Deux ouistitis communs adultes (C. jacchus) ont été utilisés pour cette expérience : Conan (homme, 2.8 ans, 0.386 kg) et Sandy (femme, 5.8 ans, 0.468 kg) (tableau supplémentaire 3 donne plus de détails). Ils ont été logés dans des conditions standard de 27°C et 50% d'humidité, avec un accès ad libitum à la nourriture et à l'eau. Tous les animaux ont été logés en groupe et les expériences ont été réalisées dans le laboratoire des systèmes corticaux et du comportement de l'UCSD. Toutes les expériences ont été approuvées par l'UCSD IACUC. La veille de l'infusion, la nourriture des animaux a été retirée.
Les animaux ont été anesthésiés avec de la kétamine (Ketaset, Zoetis 043-304, 20 mg kg-1), la peau au-dessus de la veine saphène a été rasée et désinfectée avec un gommage à l'isopropanol et 2 × 1013 vg kg-1 d'AAV a été perfusé pendant 5 min. Les animaux ont été surveillés jusqu'à ce qu'ils deviennent actifs, après quoi ils ont été remis dans leurs cages. L'activité et le comportement ont été étroitement surveillés au cours des 3 jours suivants, avec des observations quotidiennes par la suite. Des échantillons de sang ont été prélevés aux jours 1, 7, 14, 21 et 31 pour mesurer la concentration virale dans le plasma.
A 31 jours post-injection, les marmousets ont été anesthésiés avec de la kétamine comme décrit précédemment puis euthanasiés (Euthasol, Virbac 200-071, 1 ml kg-1) et perfusé avec 1 × PBS. Les cerveaux et les organes ont été coupés en deux et une moitié a été congelée rapidement dans du 2-méthylbutane (Sigma-Aldrich, M32631), refroidie avec de la neige carbonique et stockée à -80 ° C. L'autre moitié a été fixée dans du paraformaldéhyde (PFA) à 4 % (Thermo Scientific, J19943-K2) pendant une nuit, puis stockée à 4 °C dans de l'azoture de PBS (Sigma-Aldrich, S2002-100G, 0.025 %). Les échantillons ont ensuite été expédiés au California Institute of Technology (Caltech) pour analyse. Pour la coloration GLUT1, nous avons incubé des tranches avec de l'anti-GLUT1 de lapin (1:200 ; Millipore-Sigma, cat # 07-1401 ; RRID : AB_1587074), effectué trois à cinq lavages avec du PBS, incubé avec des IgG anti-lapin d'âne (1 : 200 ; Jackson ImmunoResearch Labs, cat # 711-605-152 ; RRID : AB_2492288) et lavé trois à cinq fois avant le montage. Nous avons dilué tous les anticorps et effectué toutes les incubations en utilisant du PBS additionné de 0.1 % de Triton X-100 (Sigma-Aldrich, T8787) et de 10 % de sérum d'âne normal (Jackson ImmunoResearch Labs, cat # 017-000-121 ; RRID : AB_2337258) pendant la nuit à température ambiante sous agitation.
Extraction d'ADN de bibliothèque virale et préparation d'échantillons NGS
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Nous avons précédemment signalé que l'ADN de la bibliothèque virale et l'ARN de l'hôte endogène peuvent être isolés à l'aide de TRIzol en précipitant l'acide nucléique de la phase aqueuse12,16. Par conséquent, pour extraire l'ADN de la bibliothèque virale du tissu ouistiti, nous avons homogénéisé 100 mg de moelle épinière, de foie et de chaque bloc coronal du cerveau dans du TRIzol (Life Technologies, 15596) à l'aide d'un BeadBug (Benchmark Scientific, D1036) et des acides nucléiques isolés du phase aqueuse selon le protocole préconisé par le fabricant. Nous avons traité le précipité reconstitué avec de la RNase (Invitrogen, AM2288) et digéré avec SmaI pour améliorer la récupération de l'ADN viral en aval via PCR. Après digestion, nous avons purifié avec un kit Zymo DNA Clean and Concentrator (D4033) selon le protocole recommandé par le fabricant et stocké l'ADN viral purifié à -20 ° C.
Pour ajouter des adaptateurs Illumina flanquant la région diversifiée, nous avons d'abord amplifié par PCR la région contenant notre insertion 7mer en utilisant 50% de l'ADN viral extrait total comme matrice (25 cycles). Après purification de l'ADN Zymo, nous avons dilué les échantillons à 1:100 et amplifié davantage autour de la région variable avec dix cycles de PCR, en ajoutant des régions de liaison pour la prochaine réaction de PCR. Enfin, nous avons ajouté des adaptateurs de cellule de flux Illumina et des indices uniques à l'aide d'amorces à double index NEBNext (New England Biolabs, E7600) via dix cycles supplémentaires de PCR. Nous avons ensuite purifié sur gel les produits de PCR finaux à l'aide d'un gel d'agarose à 2 % à bas point de fusion (Thermo Fisher Scientific, 16520050) et récupéré la bande de 210 pb.
Pour la bibliothèque de second tour uniquement, nous avons également isolé l'ADNsb de la bibliothèque AAV encapsidé pour NGS afin de calculer les scores d'enrichissement de la bibliothèque, une métrique quantitative que nous avons utilisée pour normaliser les différences de titre des différentes variantes de notre bibliothèque (voir réf. 16 et la section « Alignement de lecture NGS, analyse et génération de graphes de réseau »). Pour isoler les génomes viraux encapsidés, nous avons traité la bibliothèque de capsides AAV avec de la DNaseI et des capsides digérées à l'aide de la protéinase K. Nous avons ensuite purifié l'ADNsb à l'aide de phénol-chloroforme, amplifié les transgènes viraux par deux étapes d'amplification PCR pour ajouter des adaptateurs et des indices pour Illumina NGS et purifié par électrophorèse sur gel. Cet ADN de la banque virale, ainsi que l'ADN viral extrait du tissu, ont été envoyés pour un séquençage en profondeur à l'aide d'un système Illumina HiSeq 2500 (Millard et Muriel Jacobs Genetics and Genomics Laboratory, Caltech).
Alignement de lecture NGS, analyse et génération de graphes de réseau
Les fichiers FASTQ bruts des exécutions NGS ont été traités avec des scripts personnalisés (https://github.com/GradinaruLab/protfarm et de https://github.com/GradinaruLab/mCREATE)16. Pour la bibliothèque de premier tour, le pipeline de traitement de ces ensembles de données impliquait un filtrage pour supprimer les lectures de faible qualité, l'utilisation d'un score de qualité pour chaque séquence et l'élimination des biais des mutations induites par la PCR ou de la teneur élevée en GC. L'ensemble de données filtré a ensuite été aligné par un algorithme de correspondance de chaîne parfaite et ajusté pour améliorer la qualité de l'alignement. Nous avons ensuite affiché le nombre de lectures absolues pour chaque variante lors de la séquence de séquençage dans chaque tissu, et toutes les 33,314 XNUMX variantes trouvées dans le cerveau ont été choisies pour les sélections du second tour.
Après les sélections du second tour, nous avons effectué la même analyse pour afficher le nombre de lectures absolues de variantes de la bibliothèque de virus injectés et de chaque variante dans chaque tissu. De plus, nous avons calculé l'enrichissement de la bibliothèque16 pour chaque variante dans chaque tissu :
$${overline{{rm{RC}}}_{x,{rm{injecté}},{rm{bibliothèque}}}=,frac{{{rm{RC}}}_{x,{rm{ injecté}},{rm{bibliothèque}}}}{mathhop{sum }nolimits_{i=1}^{{N}_{{rm{injecté}},{rm{bibliothèque}}}}{{rm{RC }}}_{i,{rm{injecté},{bibliothèque}}}},$$
(1)
$${overline{{rm{RC}}}}_{x,{rm{tissue}}}=,frac{{{rm{RC}}}_{x,{rm{virus}}}}{mathhop {sum }nolimits_{i=1}^{{N}_{{rm{tissu}}}}{{rm{RC}}}_{i,{rm{tissu}}}},$$
(2)
$${rm{Bibliothèque},{enrichissement}}=,{log }_{10}left(frac{{overline{{rm{RC}}}}_{x,rm{{injecté},{bibliothèque}} }}{{surligne{{rm{RC}}}_{x,{rm{tissu}}}}droite),$$
(3)
telle que pour un échantillon donné y (par exemple, la banque de virus injectés ou un échantillon de tissu), RCx,y est le nombre absolu de lectures de la variante x, Ny est le nombre total de variantes récupérées et ({overline{{rm{RC}}}}_{x,{y}}) est le nombre de lectures normalisé.
Pour construire le graphique de regroupement de séquences CAP-Mac, nous avons filtré les données NGS de deuxième tour sur la base des critères suivants : (1) ≥100 nombre de lectures dans l'échantillon de bibliothèque injecté (24,186 33,314/2 0.7 variantes), (415) ≥3 enrichissement de la bibliothèque score dans plus de deux échantillons de cerveau (0.7 variantes) et (0.7) au moins deux échantillons de cerveau supplémentaires avec un enrichissement de bibliothèque ≥ 323, 2 que les échantillons de cerveau avec un enrichissement de bibliothèque inférieur à -1, 100 (2 variantes). Pour construire le graphique de séquence CAP-C0.7, nous avons filtré les données NGS de deuxième tour sur la base des critères suivants : (95) ≥ XNUMX nombre de lectures dans l'échantillon de bibliothèque injecté et (XNUMX) les deux réplicats de codons sont présents dans au moins deux échantillons de cerveau avec ≥ XNUMX enrichissement de la bibliothèque (XNUMX variantes). Ces variantes ont ensuite été traitées indépendamment pour déterminer les distances de Hamming inverses par paires (https://github.com/GradinaruLab/mCREATE) et regroupés à l'aide de Cytoscape (v. 3.9.0 ; RRID : SCR_003032) comme décrit précédemment en détail16. Les réseaux présentés montrent des variantes de capside (nœuds) reliées par des arêtes si la distance de Hamming inverse par paire est ≥3.
Clonage de variantes de capside AAV individuelles
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.n2bvj87ebgk5/v1). Pour la caractérisation à un seul variant, nous avons cloné de nouveaux plasmides variants en digérant une version modifiée du squelette pUCmini-iCAP-PHP.eB (Addgene ID : 103005 ; RRID : Addgene_103005) à l'aide de MscI et AgeI. Nous avons conçu une amorce de 100 pb qui contenait l'insertion souhaitée de 21 pb pour chaque variante de capside et les régions complémentaires de la matrice AAV9 avec ~ 20 pb chevauchant le squelette digéré. Nous avons ensuite assemblé le plasmide variant à l'aide du NEBuilder HiFi DNA Assembly Master Mix, en combinant 5 μl d'amorce de 200 nM avec 30 ng de squelette digéré dans le mélange réactionnel. Le plasmide de capside utilisé pour produire AAV.CAP-Mac est disponible sur Addgene (Addgene ID : 200658 ; RRID : Addgene_200658).
Production et purification individuelles d'AAV
Le détail de cette procédure se trouve sur protocols.io (https://doi.org/10.17504/protocols.io.14egn2dqzg5d/v1). Pour produire des variantes pour les tests en pool, nous avons suivi notre protocole précédemment publié70 en utilisant des boîtes de culture tissulaire de 150 mm. Pour la caractérisation individuelle AAV.CAP-Mac et AAV9 in vivo et in vitro, nous avons adopté notre protocole publié pour utiliser des CellSTACKs à dix couches (Corning, 3320) pour produire efficacement des virus à un titre élevé pour doser les macaques rhésus et les singes verts. Plus précisément, nous avons passé vingt plats de 150 mm à environ 70 % de confluence dans un CellSTACK à dix couches 24 h avant la transfection. Le jour de la transfection, nous avons préparé le mélange de transfection PEI-ADN pour quarante boîtes de 150 mm et combiné le mélange de transfection avec du milieu et effectué un changement complet de milieu pour le CellSTACK. Nous avons collecté et changé le support à 72 h post-transfection similaire à la production dans des boîtes de 150 mm. 120 h après la transfection, nous avons ajouté de l'acide éthylènediaminetétraacétique (Invitrogen, 15575020) à une concentration finale de 10 mM et incubé à 37 ° C pendant 20 min, en remuant et en tapotant occasionnellement les côtés du CellSTACK pour détacher les cellules. Nous avons ensuite retiré le mélange de médias et de cellules et procédé au protocole de purification AAV70. Il convient de noter que lors de l'étape d'échange de tampon après ultracentifugation, nous avons utilisé des concentrateurs de protéines centrifuges avec des membranes en polyéthersulfone (Thermo Scientific, 88533) au lieu d'appareils de filtration Amicon et utilisé du PBS de Dulbecco additionné de 0.001 % de Pluronic F-68 (Gibco, 24040032).
Expériences sur les rongeurs
Toutes les procédures de rongeurs ont été effectuées à Caltech et ont été approuvées par l'IACUC local. Nous avons acheté des souris C57BL/6J (souche # : 000664 ; RRID : IMSR_JAX : 000664), BALB/cJ (souche # : 000651 ; RRID : IMSR_JAX : 000651) et DBA/2J (souche # : 000671 ; RRID : IMSR_JAX : 000671) (tous des mâles, âgés de 6 à 8 semaines) du Jackson Laboratory. Pour l'administration IV chez la souris, nous avons délivré 5 × 1011 vg de virus à travers le sinus rétro-orbitaire70,71 à l'aide d'une seringue à insuline de calibre 31 (BD, 328438). Voir protocols.io pour plus de détails sur les injections rétro-orbitales d'AAV chez la souris (https://doi.org/10.17504/protocols.io.3byl4joy8lo5/v1). Pour l'administration intracérébroventriculaire (ICV) chez la souris, nous avons injecté 5.0 × 1010 ou 1.5 × 1011 vg dans le ventricule latéral. En bref, nous avons anesthésié des souris à l'aide d'isoflurane (5 % pour l'induction, 1 à 3 % pour l'entretien) avec 95 % d'O2/ 5% CO2 (1 litre min-1) et les souris ont été fixées à la tête dans un cadre stéréotaxique. Après avoir rasé la tête et stérilisé la zone avec de la chlorohexidine, nous avons administré par voie sous-cutanée 0.05 ml de 2.5 mg ml-1 bupivacaïne, et une incision médiane a été faite et le crâne a été nettoyé du sang et du tissu conjonctif. Après avoir nivelé la tête, des trous de fraisage ont été percés bilatéralement au-dessus des ventricules latéraux (0.60 mm en arrière du bregma et à 1.15 mm de la ligne médiane). Les vecteurs viraux ont été aspirés dans des seringues NanoFil de 10 µl (World Precision Instruments) à l'aide d'une aiguille de microinjection de calibre 33, et l'aiguille a été lentement abaissée dans le ventricule latéral (à 1.6 mm de la surface piale). L'aiguille a été laissée en place pendant environ 5 min et 3 à 5 µl de vecteur viral ont été injectés à l'aide d'une pompe à microseringue (World Precision Instruments, UMP3) et d'un contrôleur de pompe (World Precision Instruments, Mircro3) à un débit de 300 nl min-1. Toutes les souris ont reçu en peropératoire 1 mg kg-1 de buprénorphine LP et 5 mg kg-1 de kétoprofène par voie sous-cutanée et 30 mg kg-1 d'ibuprofène et 60 mg kg-1 de triméthoprime/sulfaméthoxazole pendant 5 jours après la chirurgie. Voir protocols.io pour plus de détails sur les injections ICV d'AAV chez la souris (https://doi.org/10.17504/protocols.io.5qpvorm4dv4o/v1). Après 3 semaines d'expression, toutes les souris ont été perfusées avec du PBS et fixées dans du PFA à 4 %. Tous les organes ont été extraits, incubés dans du PFA à 4.00 % pendant la nuit, transférés dans du PBS additionné d'azide de sodium à 0.01 % et stockés à 4 °C pour un stockage à long terme. Nous avons découpé le cerveau en sections de 100 μm avec un vibratome (Leica Biosystems, VT1200S), monté dans Prolong Diamond Antifade (Invitrogen, P36970) et imagé à l'aide d'un microscope confocal (Zeiss, LSM 880) en utilisant ZEN (édition noire). Voir protocols.io pour plus de détails sur la manipulation des tissus (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 et de https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1).
Expériences sur les macaques rhésus
Les détails des procédures de cette section peuvent être trouvés sur protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1). Les macaques nouveau-nés (0.45 à 1.40 kg) ont été sevrés à la naissance. Au cours du premier mois, les macaques ont reçu des vecteurs AAV par voie intraveineuse ou intrathécale. Tous les macaques adultes (8 à 17 ans ; 4.65 à 11 kg) inclus dans cette étude ont reçu une perfusion d'AAV par voie intraveineuse uniquement. Pour les injections IV, les animaux ont été anesthésiés avec de la kétamine (0.10 ml) et la peau au-dessus de la veine saphène a été rasée et désinfectée. AAV (entre 2 × 1013 et 1 × 1014 vg kg-1) a été lentement infusé dans la veine saphène pendant environ 1 min dans <0.75 ml de PBS. Pour les injections d'ICM, les animaux ont reçu un sédatif par voie intramusculaire et la zone de la peau au niveau du cou a été rasée et préparée aseptiquement. Une aiguille a été avancée dans la citerne magna pour retirer une petite quantité de LCR proportionnelle à la quantité de liquide injectée. Ensuite, une seringue stérile contenant la préparation stérile de l'AAV (1.5 × 1012 ou 2.5 × 1013 vg kg-1) proportionnel à la quantité de liquide collecté a été fixé de manière aseptique et injecté lentement. Tous les animaux ont été surveillés pendant la récupération de la sédation tout au long de la journée, puis quotidiennement pour tout résultat indésirable. Tous les singes ont été logés individuellement à la vue et au son des congénères. Les tissus ont été prélevés 4 à 11 semaines après l'injection. Les animaux ont été profondément anesthésiés et ont reçu du pentobarbital de sodium conformément aux directives pour l'euthanasie sans cruauté des animaux au California National Primate Research Center. Tout le matériel injecté aux macaques rhésus était exempt d'endotoxines (<0.1 EU ml-1), et la pureté des protéines a été confirmée par électrophorèse sur gel de dodécylsulfate de sodium et de polyacrylamide. Tableaux supplémentaires 4 et de 5 répertorier la voie d'administration, les variantes d'AAV, la dose virale, la cargaison génétique et la durée d'expression pour chaque expérience.
Tests en piscine chez les macaques rhésus
Les détails des procédures de cette section peuvent être trouvés sur protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1, https://doi.org/10.17504/protocols.io.3byl4jo68lo5/v1 et de https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Des expériences de bassin de macaques nouveau-nés (RM-001 à RM-004) ont été réalisées au CNPRC de l'UC Davis et approuvées par l'IACUC local. Des expériences de piscine de macaques adultes (RMN-001 et RMN-002) ont été réalisées au NIMH et approuvées par leur IACUC local. Les macaques ont été perfusés avec du PBS glacé sans RNase. Au moment de la perfusion, un hémisphère du cerveau a été congelé instantanément et l'autre hémisphère a été sectionné en blocs coronaux de 4 mm et post-fixé dans du PFA à 4 % pendant 48 h et transféré à Caltech pour un traitement ultérieur. Pour la coloration HA, nous avons incubé des tranches avec du lapin anti-HA (1:200 ; Cell Signaling Technology, cat # 3724 ; RRID : AB_1549585), effectué trois à cinq lavages avec du PBS, incubé avec des IgG anti-lapin d'âne (1:200 ; Jackson ImmunoResearch Labs, cat # 711-605-152 ; RRID : AB_2492288) et lavé trois à cinq fois avant le montage. Nous avons dilué tous les anticorps et effectué toutes les incubations en utilisant du PBS additionné de 0.1 % de Triton X-100 (Sigma-Aldrich, T8787) et de 10 % de sérum d'âne normal (Jackson ImmunoResearch Labs, cat # 017-000-121 ; RRID : AB_2337258) une nuit à température ambiante sous agitation.
Pour isoler l'ADN viral et l'ARN entier, des tranches de 100 mg de cerveau et de foie ont été homogénéisées dans du TRIzol (Life Technologies, 15596) à l'aide d'un BeadBug (Benchmark Scientific, D1036) et l'ADN et l'ARN totaux ont été récupérés selon le protocole recommandé par le fabricant. . L'ADN récupéré a été traité avec de la RNase, digéré par restriction avec Smal et purifié avec un kit Zymo DNA Clean and Concentrator (D4033). L'ARN récupéré a été traité avec de la DNase, et l'ADNc a été généré à partir de l'ARNm en utilisant SuperScript III (Thermo Fisher Scientific, 18080093) et des amorces oligo(dT) selon le protocole recommandé par le fabricant. Nous avons utilisé la PCR pour amplifier la région du code-barres en utilisant 50 ng d'ADN viral ou d'ADNc comme matrice. Après la purification de l'ADN Zymo, nous avons dilué les échantillons à 1:100 et amplifié davantage la région du code-barres à l'aide d'amorces pour ajouter des adaptateurs pour Illumina NGS. Après nettoyage, ces produits ont été encore amplifiés à l'aide d'amorces à double index NEBNext pour le séquençage Illumina (New England Biolabs, E7600) pendant dix cycles. Nous avons ensuite purifié sur gel les produits PCR finaux en utilisant un gel d'agarose à 2 % à bas point de fusion (Thermo Fisher Scientific, 16520050). L'enrichissement des tests de pool a été calculé de manière identique à l'enrichissement de la bibliothèque, mais est représenté à la Fig. 2b, c sur une échelle linéaire.
Caractérisation individuelle de CAP-Mac chez les macaques rhésus
Les détails des procédures de cette section peuvent être trouvés sur protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 et de https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Des macaques nouveau-nés ont été perfusés avec du PBS et du PFA à 4 %. Le cerveau a été sectionné en blocs coronaux de 4 mm et tout le tissu a été post-fixé dans du PFA à 4 % pendant 3 jours avant stockage dans du PBS. Le macaque adulte unique utilisé pour la caractérisation individuelle in vivo (RM-020; 17 ans, 11 kg) a été perfusé avec du PBS sans RNase, et un demi-hémisphère a été congelé instantanément et l'autre sectionné en blocs coronaux de 4 mm et post-fixé dans 4% PFA. Tous les tissus ont été transférés à Caltech pour un traitement ultérieur. Les cerveaux et les foies ont été sectionnés en tranches de 100 μm à l'aide d'un vibratome. De plus, des sections de cerveau et de moelle épinière ont été incubées dans du saccharose à 30% pendant une nuit et incorporées dans un composé OCT (Scigen, 4586) et sectionnées en tranches de 50 μm à l'aide d'un cryostat (Leica Biosystems, CM1950). Toutes les tranches ont été montées à l'aide de Prolong Diamond Antifade et imagées à l'aide d'un microscope confocal. Pour la coloration GFP de la moelle épinière et des tranches de cerveau du macaque administré par voie intrathécale, nous avons incubé des tranches avec du poulet anti-GFP (1:500 ; Aves Labs, cat # GFP-1020 ; RRID : AB_10000240), effectué trois à cinq lavages avec du PBS , incubé avec IgY anti-poulet d'âne (1:200 ; Jackson ImmunoResearch Lab, cat # 703-605-155 ; RRID : AB_2340379) et lavé trois à cinq fois avant le montage. Nous avons dilué tous les anticorps et effectué toutes les incubations en utilisant du PBS additionné de 0.1 % de Triton X-100 (Sigma-Aldrich, T8787) et de 10 % de sérum d'âne normal (Jackson ImmunoResearch, 017-000-121) pendant une nuit à température ambiante sous agitation.
Pour la reconstruction morphologique, nous avons sectionné les cerveaux en sections de 300 μm et les avons incubés dans une solution d'adaptation d'indice de réfraction72 pendant 72 h avant de monter sur une lame immergée dans la solution d'adaptation d'indice de réfraction. Nous avons imagé à l'aide d'un microscope confocal et d'un objectif × 25 (LD LCI Plan-Apochromat × 25 / 0.8, 100 Imm Corr DIC) en utilisant XNUMX% de glycérol comme fluide d'immersion. Nous avons capturé du carrelage Z piles (1,024 1,024 × 007370 XNUMX pour chaque image en utilisant les paramètres de capture suggérés) autour des cellules d'intérêt et des champs de vision appropriés recadrés pour le traçage. Le traçage a été effectué dans Imaris (Oxford Instruments ; RRID : SCR_XNUMX) en utilisant des méthodes semi-automatisées et automatisées.
Pour la quantification des neurones (NeuN) et des astrocytes (S100β), les tranches ont été colorées à l'aide d'un anticorps anti-NeuN (EPR12763) (1:200 ; Abcam, cat # ab177487 ; RRID : AB_2532109) ou d'un anticorps anti-S100β (1:200 ; Abcam , cat # ab52642 ; RRID : AB_882426) pendant une nuit dans du PBS additionné de 0.1 % de Triton X-100 et de 10 % de sérum d'âne normal. Les tranches ont été lavées trois à cinq fois avec du PBS et incubées pendant une nuit dans un anticorps IgG anti-lapin conjugué avec Alexa Fluor 647 (1:200 ; Jackson ImmunoResearch Labs, cat # 711-605-152 ; RRID : AB_2492288) dans du PBS + 0.1 % Triton X-100 + 10% de sérum d'âne normal. Après trois à cinq lavages et montage avec Prolong Diamond Antifade, nous avons obtenu Z empilements à l'aide d'un microscope confocal et d'un objectif x25. Nous avons segmenté les cellules positives NeuN et XFP à l'aide de scripts personnalisés en Python (RRID : SCR_008394) et Cellpose (https://www.cellpose.org/; RRID : SCR_021716)73.
Imagerie à deux photons ex vivo
Des tranches de cerveau de tailles adaptées à l'imagerie ont été préparées avec une épaisseur de 400 µm à partir de tranches plus grandes à l'aide d'un vibratome et stockées dans du liquide céphalo-rachidien artificiel barboté avec du gaz carbogène avant l'imagerie à deux photons, comme décrit précédemment dans les protocoles publiés74,75. Pour tester les réponses GCaMP8s, une stimulation électrique (4–5 V, 80 Hz, durée 0.3 s) avec le nombre d'impulsions indiqué a été délivrée à l'aide d'une électrode monopolaire extracellulaire placée à 100–200 µm du neurone imagé. La fréquence d'image de l'imagerie était de 30 Hz. Les traces de régions d'intérêt segmentées ont été tracées en tant que ΔF/F0 = (F(t) − F0)/F0, Où F0 est défini comme la moyenne de toutes les valeurs de fluorescence avant la stimulation électrique. Le temps de montée a été défini comme le temps nécessaire à la phase montante du signal pour passer de 10% du pic à 90% du pic. La constante de temps de décroissance a été obtenue en ajustant les sommes des exponentielles à la phase de décroissance du signal. Le rapport signal sur bruit a été obtenu en divisant l'amplitude du pic du signal par l'écart type de la trace de fluorescence avant la stimulation électrique.
Caractérisation en tranche de macaque rhésus adulte
Un macaque rhésus adulte (14 ans et 1 mois; 10.83 kg) du Washington National Primate Research Center devait être euthanasie de routine, et le cerveau a été prélevé dans le cadre du programme de distribution de tissus de l'établissement. Un bloc du gyrus temporal supérieur a été sectionné en tranches de 300 μm et les tranches ont été récupérées74 et cultivé sur une interface air-membrane liquide, comme décrit précédemment76. Environ 30 min après avoir étalé les tranches, nous avons administré 1 à 2 μl d'AAV (5 × 1013 vg ml-1 de l'emballage AAV9 ou AAV.CAP-Mac soit ssCAG-FXN-HA ou ssCAG-eGFP). Les expériences ont été réalisées en triplicats biologiques pour chaque condition et le milieu de culture a été renouvelé toutes les 48 h jusqu'à la collecte des tissus à 8 jours post-transduction. Le jour de la collecte des tissus, les tranches ont été imagées pour confirmer la transduction, les tranches ont été coupées en deux et chaque demi-tranche a été congelée instantanément dans un bain de glace sèche et d'éthanol. Les échantillons ont été stockés à -20 ° C jusqu'à ce qu'ils soient traités ultérieurement.
Chaque demi-tranche a été traitée (une pour la récupération d'ADN et d'ARN). L'ADN a été isolé à l'aide du kit Qiagen DNeasy Blood and Tissue (Qiagen, catalogue n° 69504) et l'ARN a été récupéré à l'aide de TRIzol (Thermo Fisher Scientific, catalogue n° 15596026) et du kit PureLink RNA Mini (Thermo Fisher Scientific, catalogue n° 12183018A). L'ADN a été retiré de l'échantillon d'ARN en modifiant le premier lavage du kit PureLink RNA Mini comme suit : laver avec 350 µl de tampon de lavage 1, puis ajouter 80 µl de DNaseI sans RNase dans du tampon RDD (catalogue Qiagen # 79254) et incuber la colonne à température ambiante pendant 15 min ; puis laver à nouveau avec 350 µl de tampon de lavage 1 avant de poursuivre le protocole. Nous avons effectué la synthèse d'ADNc du premier brin à partir de 400 ng d'ARN total dans des réactions de 20 µl à l'aide d'un kit Promega GoScript Reverse Transcription (Promega, catalogue # A5000).
Nous avons ensuite évalué les génomes des vecteurs et les transcrits viraux trouvés dans chaque échantillon à l'aide d'une PCR quantitative sur un Roche Lightcycler II. Ici, 100 ng d'ADN ont été utilisés dans une réaction d'amplification de 20 µl à l'aide de sondes TaqMan de Thermo Fisher Scientific (sonde EGFP-FAM, essai ID Mr04097229_mr, catalogue #4331182 ; sonde de référence génomique personnalisée CN2386-2-VIC, essai ID ARH6DUK, catalogue # 4448512, conçu pour cibler à la fois M. mulâtre et de Macaca Nemestrina).
Expériences de singe vert
Tout le singe vert (C. sabaeus) les procédures ont été réalisées à Virscio et approuvées par leur IACUC. Tous les singes ont été criblés pour les anticorps neutralisants et confirmés comme ayant un titre <1:5. À environ 7 à 8 mois (1.0 à 1.3 kg), les singes ont reçu une dose intraveineuse (tableau supplémentaire 6). Les formulations de dose ont été laissées s'équilibrer à environ la température ambiante pendant au moins 10 min, mais pas plus de 60 min avant le dosage. Les volumes de dose IV étaient basés sur les poids corporels au jour 0. Les animaux ont été sédatés avec de la kétamine (8.0 mg kg-1) et xylazine (1.6 mg kg-1). La zone d'injection a été rasée et préparée avec de la chlorohexidine et de l'isopropanol à 70 % et nettoyée chirurgicalement avant l'insertion du cathéter IV. Le dosage a eu lieu avec une seule perfusion IV d'AAV (7.5 × 1013 ou 7.6 × 1013 vg kg-1) au jour 0 via la veine saphène administré à l'aide d'un dispositif de perfusion portatif à un débit cible de 1 ml min-1. Le bien-être général a été confirmé deux fois par jour par une observation en cage commençant 1 semaine avant l'administration. Au moment prévu de l'euthanasie, les singes ont été mis sous sédation avec de la kétamine (8 à 10 mg kg-1 intramusculaire) et euthanasié avec du pentobarbital sodique (100 mg kg-1 IV à effet). En cas de perte du réflexe cornéen, une perfusion transcardiaque (ventricule gauche) a été réalisée avec du PBS refroidi à l'aide d'une pompe péristaltique réglée à un débit d'environ 100 ml min-1 jusqu'à ce que le liquide qui s'échappe soit clair avant le prélèvement des tissus. Des cubes de tissu ont été prélevés dans l'hémisphère gauche du cerveau et divers autres organes et congelés dans la phase vapeur d'azote liquide pour un traitement ultérieur pour la biodistribution. L'hémisphère droit du cerveau a été retiré et coupé en tranches coronales d'environ 4 mm et post-fixé intact avec environ 20 volumes de formol tamponné neutre à 10 % pendant environ 24 h à température ambiante.
L'ADN génomique a été extrait du SNC et des tissus périphériques à l'aide du kit d'extraction Thermo Fisher MagMax DNA Ultra 2.0 (catalogue # A36570). Le rendement de l'ADN a été évalué par quantification fluorométrique avec le test d'ADNdb Qubit. Environ 20 ng d'ADN ont été chargés dans chaque réaction de 20 μl et les plaques ont été passées sur le système de détection par PCR en temps réel BioRad CFX Connect (catalogue # 1855201). Le dosage du nombre de copies virales a été validé pour la spécificité par la détection d'un seul produit amplifié ; la sensibilité, en évaluant la limite inférieure de détection à plus de dix copies par réaction ; et linéarité, en assurant la courbe standard R2 était > 0.95. Les réactions ont été assemblées dans FastStart Universal SYBR Green Master (Rox) (catalogue # 4913850001). Les séquences des amorces étaient ACGACTTCTTCAAGTCCGCC (sens) et TCTTGTAGTTGCCGTCGTCC (sens inverse). Le protocole PCR a utilisé une étape de dénaturation initiale de 95 ° C pendant 180 s, suivie de 40 cycles de 95 ° C pendant 15 s et de 60 ° C pendant 60 s, avec une étape d'imagerie après chaque cycle de 60 ° C. Une courbe standard a été générée avec un plasmide linéarisé contenant la séquence de matrice GFP présente dans le virus à partir de 1 × 108 à 1 × 1010 copies, diluées dans des échantillons d'ADN de macaque naïfs non traités préparés à l'aide d'un kit identique à celui des échantillons de cette étude pour contrôler les effets de matrice. Les copies d'ADN viral ont été calculées à partir de la courbe standard en utilisant l'équation de la ligne de meilleur ajustement. La multiplicité des valeurs d'infection a été calculée sur la base du poids génomique total mesuré de l'ADN de la cellule hôte par réaction.
Après la fixation, les tissus ont été placés dans 10 % > 20 % > 30 % de saccharose pendant 24 h chacun à 4 °C, puis intégrés dans un composé OCT et stockés à -80 °C jusqu'à la cryosection. Les blocs de tissus ont été portés à -20 ° C dans un cryostat avant d'être sectionnés en tranches de 30 μm et montés à sec sur des lames après cryosection. Après sectionnement, les lames ont été laissées à température ambiante pendant une nuit pour sécher. Pour aider à la quantification des neurones, nous avons coloré les coupes avec les anticorps et concentrations suivants : lapin anti-GFP (1:100 ; Millipore-Sigma, cat # AB3080 ; RRID : AB_91337) et souris anti-NeuN (A60) (1:500 ; Millipore-Sigma, cat # MAB377 ; RRID : AB_2298772). Pour la coloration des anticorps secondaires, les anticorps secondaires et concentrations suivants ont été utilisés : âne anti-lapin Alexa Fluor 488 (1:500 ; Thermo Fisher Scientific, cat # A-21206 ; RRID : AB_2535792) et âne anti-souris Alexa Fluor 647 (1 :500 ; Thermo Fisher Scientific, numéro de catalogue A-31571 ; RRID : AB_162542). Tous les anticorps ont été dilués avec du PBS 1X additionné de 0.25 % de Triton X-100 (PBST) et de 5.00 % de sérum d'âne normal. Les incubations d'anticorps primaires ont été laissées une nuit à température ambiante. Les sections ont ensuite été lavées avec du PBST. Des incubations d'anticorps secondaires ont été réalisées pendant 2 h à température ambiante. Les coupes ont été lavées trois fois dans du PBST. Les coupes ont été incubées dans une solution DAPI (1:10,000 1306 ; Invitrogen, D5) à température ambiante pendant XNUMX min puis lavées. Les coupes ont été recouvertes à l'aide de Prolong Diamond Antifade.
Trois coupes par animal ont été colorées et imagées. Chaque section a été imagée en triple, chaque région d'intérêt ayant un total de neuf images. Les régions tissulaires d'intérêt ont été imagées avec un Keyence BZ-X800 avec les paramètres d'acquisition suivants : GFP (1/500 s), Cy5 (1 s), DAPI (1/12 s), haute résolution Z pile au pas de 1.2 µm. Les sous-régions cérébrales suivantes ont été imagées : cortex frontal, pariétal, temporal, occipital, cervelet, caudé, putamen et thalamus (noyaux médial, ventral latéral et ventral postérieur). Une méthode semi-automatisée de comptage de cellules a été réalisée à l'aide d'ImageJ (RRID : SCR_003070) pour la quantification. À l'aide de seuils et d'analyses de particules, nous avons quantifié les cellules NeuN-positives et DAPI-positives. À l'aide du compteur de cellules d'ImageJ, nous avons compté manuellement les cellules GFP-positives ainsi que les cellules GFP et NeuN double-positives.
Expériences iPSC
Des cultures neuronales ont été produites en différenciant et en faisant mûrir des cellules progénitrices neurales dérivées d'iPSC avec des kits de différenciation et de maturation du cerveau antérieur Stemdiff (StemCell # 08600 et # 08605, respectivement), conformément aux protocoles de leur fabricant. Des cellules progénitrices neurales ont été produites par différenciation de la lignée iPSC dérivée de fibroblastes de prépuce : ACS-1019 (ATCC # DYS-0100 ; RRID : CVCL_X499), avec les kits Stemdiff SMADi Neural Induction (StemCell l#08581), sélection avec Stemdiff Neural Rosette Selection Réactif (StemCell l#05832) et expansion dans Stemdiff Neural Progenitor Media (StemCell l#05833), selon les protocoles de leur fabricant. Les neurones ont été mûris un minimum de 8 jours avant le replaquage pour la transduction.
Des cultures neuronales matures, ensemencées à 15,000 96 cellules par puits dans des plaques optiques à 4 puits à parois noires revêtues de polyornithine et de laminine, ont été cultivées pendant 90 jours supplémentaires avant la transduction. Des puits répliqués ont été transduits avec du virus dilué en série sur six ordres de grandeur dans un milieu de maturation à 10 % et 4 % d'OptiPRO SFM. Quatre jours après la transduction, les cultures ont été fixées avec 1 % de PFA et contre-colorées avec XNUMX µg ml-1 Hoechst 33322. L'identification des cellules transduites a été déterminée par imagerie de 60 champs par puits, en utilisant une détection par fluorescence à deux canaux (Hoechst, ex386/em440 ; eGFP, ex485/em521) sur une plate-forme CellInsight CX5 HCS. Les cellules individuelles ont été identifiées par la détection Hoechst de leurs noyaux et l'application de masques annulaires de taille et de contact contraints à chaque cellule. La transduction cellulaire a été déterminée en mesurant la fluorescence eGFP au-dessus d'un niveau seuil dans un masque annulaire individuel. Pour chaque population, le pourcentage de cellules transduites a été tracé en fonction de la dose appliquée. Ajustements de courbe et CE50 les valeurs ont été déterminées avec la méthode de régression de l'agoniste Prism GraphPad (RRID : SCR_002798) par rapport à la réponse (trois paramètres). Pour rendre compte des efficacités d'expression de l'eGFP par cellule, les intensités de fluorescence des points eGFP ont été moyennées à partir de chaque masque annulaire sur un minimum de 5,000 XNUMX cellules par puits. Les ajustements de courbe ont été obtenus en utilisant le Prism GraphPad Biphasic X comme méthode de régression de concentration.
Statistiques et reproductibilité
Pour les images représentatives, au moins trois tranches distinctes de chaque échantillon ont été montées pour l'imagerie. Dans chaque région du cerveau d'un seul animal, au moins trois champs de vision différents ont été pris (champ de vision minimum après carrelage, 2.38 mm × 2.38 mm; épaisseur de tranche, 50 µm), ce qui équivaut à neuf champs de vision distincts sur trois tranches de cerveau , pour assurer la cohérence entre les échantillons d'imagerie.
Résumé du rapport
De plus amples informations sur le design de la recherche sont disponibles dans Sommaire des rapports sur le portefeuille de la nature lié à cet article.
- Contenu propulsé par le référencement et distribution de relations publiques. Soyez amplifié aujourd'hui.
- PlatoData.Network Ai générative verticale. Autonomisez-vous. Accéder ici.
- PlatoAiStream. Intelligence Web3. Connaissance Amplifiée. Accéder ici.
- PlatonESG. Automobile / VE, Carbone, Technologie propre, Énergie, Environnement, Solaire, La gestion des déchets. Accéder ici.
- Décalages de bloc. Modernisation de la propriété des compensations environnementales. Accéder ici.
- La source: https://www.nature.com/articles/s41565-023-01419-x
- :est
- :où
- $UP
- 000
- 1
- 10
- 100
- 11
- 12
- 13
- 14
- 15%
- 16
- 17
- 180
- 2%
- 20
- 200
- 2011
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 24
- 25
- 27
- 30
- 300
- 31
- 33
- 39
- 40
- 50
- 500
- 60
- 66
- 7
- 70
- 72
- 75
- 8
- 80
- 9
- 95%
- a
- au dessus de
- Absolute
- accès
- conformité
- Selon
- atteindre
- acquisition
- à travers
- infection
- activité
- Ad
- ajouter
- ajoutée
- Supplémentaire
- En outre
- administré
- administration
- adopté
- Adulte
- Avancée
- négatif
- Après
- encore
- âge
- AL
- Alexa
- algorithme
- aligné
- Tous
- permis
- le long de
- aussi
- montant
- Amplification
- Amplified
- an
- selon une analyse de l’Université de Princeton
- Présentatrice
- et de
- animal
- animaux
- Anticorps
- tous
- appliqué
- Application
- approprié
- ,
- d'environ
- Réservé
- autour
- article
- artificiel
- AS
- assemblé
- Assemblée
- évalué
- Évaluation
- aider
- At
- Automatisation
- disponibles
- moyen
- et
- Colonne vertébrale
- BANDE
- basé
- BD
- BE
- est devenu
- before
- Début
- va
- référence
- LES MEILLEURS
- jusqu'à XNUMX fois
- biais
- propriétés de liant
- Noir
- Block
- Blocs
- sang
- corps
- né
- tous les deux
- BP
- Cerveau
- cerveaux
- brièvement
- Apporté
- tampon
- mais
- by
- cages
- calculer
- calculé
- Californie
- CAN
- capturer
- capturé
- les soins
- Cargaison
- CHAT
- cellule
- Cellules
- cellulaire
- Canaux centraux
- central
- chaîne
- chan
- Change
- modifié
- choisi
- clair
- cliquez
- étroitement
- regroupement
- collection
- Colonne
- combiné
- combinant
- comité
- Commun
- complémentaire
- complet
- Complété
- conformité
- se conformer
- Composé
- concentration
- condition
- conditions
- Confirmer
- CONFIRMÉ
- NOUS CONTACTER
- connecté
- constant
- construire
- contenu
- contient
- des bactéries
- contrôleur
- copies
- Core
- coronaire
- Counter
- critères
- Culture
- courbe
- Customiser
- Construit sur mesure
- Cut/Taille
- cycle
- cycles
- Tous les jours
- données
- ensembles de données
- Davis
- journée
- jours
- profond
- défini
- livré
- page de livraison.
- décrit
- Conception
- un
- voulu
- détails
- Détection
- Déterminer
- déterminé
- déviation
- dispositif
- Compatibles
- Diamant
- Diego
- différences
- différent
- dilution
- Commande
- dans
- distance
- distinct
- distribution
- diversifié
- Diversité
- adn
- fait
- dose
- dosage
- motivation
- sécher
- durée
- pendant
- e
- E & T
- chacun
- Plus tôt
- édition
- effet
- les effets
- efficacité
- efficace
- efficacement
- non plus
- éléments
- l'élimination
- intégré
- de l'Angleterre
- assurer
- assurer
- établies
- Ether (ETH)
- EU
- évalué
- Chaque
- exemple
- échange
- avec des données
- expérience
- expériences
- expression
- extrait
- extraction
- famille
- Fed
- femelle
- champ
- Des champs
- figues
- Figure
- Fichiers
- filtration
- finale
- finalement
- résultats
- Prénom
- s'adapter
- raccord
- cinq
- fixé
- Flash
- flux
- fluide
- suivi
- Abonnement
- suit
- nourriture
- Pour
- pour le rendement
- Avant
- trouvé
- quatre
- CADRE
- gratuitement ici
- De
- gelé
- fonctionnel
- plus
- GAS
- jauge
- Général
- générer
- généré
- génération
- Génétique
- génome
- génomique
- donné
- graphique
- plus grand
- Vert
- Réservation de groupe
- Groupes
- lignes directrices
- Half
- Maniabilité
- Vous avez
- ayant
- front
- Santé
- ici
- Haute
- haute résolution
- des trous
- homogénéisé
- hôte
- HTTPS
- humain
- VÉLO
- ID
- identique
- Identification
- identifié
- if
- ii
- iii
- satellite
- Imagerie
- immergé
- immersion
- améliorer
- in
- inclus
- incubé
- indépendamment
- indice
- indiqué
- Indices
- individuel
- Individuellement
- induction
- infection
- d'information
- infusé
- infusion
- initiale
- possible
- plutôt ;
- Institut
- DOCUMENTS
- instruments
- intérêt
- Interfaces
- développement
- intraveineux
- introduit
- impliqué
- isolé
- Jackson
- kit
- laboratoire
- laboratoire
- Labs
- plus importantes
- ld
- au
- à gauche
- moins
- Niveau
- bibliothèques
- Bibliothèque
- VIE
- LIMIT
- Gamme
- LINK
- lié
- Liquide
- Liste
- Foie
- locales
- long-term
- perte
- baisser
- réduit
- LES PLANTES
- facile
- manuellement
- cartographie
- masque
- Masques
- maître
- Match
- assorti
- Matériel
- Matrice
- mesurer
- mesuré
- mesure
- Médias
- moyenne
- mental
- La santé mentale
- méthode
- méthodes
- métrique
- souris
- Microscope
- m.
- minimum
- minutes
- mélanger
- mélange
- ML
- modifié
- surveillé
- Mois
- mois
- PLUS
- ARNm
- MSCI
- à savoir
- nanotechnologies
- Nationales
- Nature
- Près
- nécessaire
- réseau et
- réseaux
- Neural
- neuronale
- Neurones
- Nouveauté
- next
- NIH
- aucune
- nœuds
- Ordinaire
- nombre
- objectif
- obtenu
- a eu lieu
- of
- Vieux
- on
- ONE
- uniquement
- optimaux
- optimisé
- or
- passer commande
- Autre
- nos
- plus de
- du jour au lendemain
- Oxford
- Oxygène
- l'emballage
- apparié
- paramètres
- partie
- particule
- PBS
- PCR
- Courant
- Penn
- Pennsylvanie
- /
- pourcentage
- parfaite
- effectué
- périphérique
- phase
- pipeline
- Emplacement
- Place
- prévu
- Plasma
- plateforme
- Platon
- Intelligence des données Platon
- PlatonDonnées
- plus
- pool
- population
- portefeuille
- La précision
- préparation
- préparé
- représentent
- présenté
- empêcher
- précédemment
- primaire
- apprêt
- sonde
- procédure
- procédures
- processus
- traité
- traitement
- produire
- Produit
- Produit
- Vidéo
- Produits
- ancêtre
- Programme
- Protecteur
- Protéines
- Protéines
- protocole
- protocoles
- fournit
- publié
- pompe
- acheté
- Python
- qualité
- quantitatif
- Qubit
- Lapin
- collectés
- Tarif
- rapport
- nous joindre
- réaction
- réactions
- Lire
- en temps réel
- reçu
- recommandé
- récupération
- région
- régions
- régression
- régulateurs
- relatif
- supprimez
- Supprimé
- réplique
- rapport
- Signalé
- Rapports
- représentant
- représenté
- conditions
- un article
- respectivement
- réponse
- réponses
- restriction
- résultant
- inverser
- bon
- Bagues
- Augmenter
- hausse
- ARN
- robuste
- roche
- Salle
- Itinéraire
- Courir
- fonctionne
- s
- même
- San
- San Diego
- sablonneux
- Escaliers intérieurs
- prévu
- SCI
- sur une base scientifique
- But
- scores
- dépistage
- scripts
- secondaire
- Section
- les sections
- sur le lien
- segmentation
- choisi
- sélection
- sélectif
- Sensibilité
- envoyé
- séparé
- Séquence
- séquençage
- Sérum
- set
- Paramétres
- plusieurs
- expédiés
- montrer
- Accompagnements
- Vue
- Signal
- similaires
- unique
- s'asseoir
- SIX
- tailles
- Peau
- Tranche
- Glissement
- Mules
- Lentement
- petit
- sodium
- sur mesure
- Son
- spécifiquement
- spécificité
- Spot
- empiler
- Combos
- Standard
- étapes
- Étapes
- storage
- stockée
- Chaîne
- Étude
- ultérieur
- convient
- haut
- Surface
- haute
- combustion propre
- systémique
- Système
- table
- tâches
- tapotement
- Target
- des campagnes marketing ciblées,
- ciblage
- Les technologies
- Technologie
- modèle
- Dix
- terminal
- Essais
- que
- qui
- La
- La Région
- leur
- Les
- puis
- Là.
- donc
- Ces
- l'ont
- this
- trois
- порог
- Avec
- tout au long de
- fiable
- fois
- tissus
- à
- a
- Total
- tracer
- Traçant
- transférer
- transféré
- traités
- Triton
- Twice
- torsion
- deux
- types
- Ultra
- sous
- expérience unique et authentique
- Universel
- université
- Université de la Californie
- Université de Pennsylvanie
- jusqu'à
- utilisé
- d'utiliser
- en utilisant
- utiliser
- Utilisant
- validé
- Valeurs
- Variante
- divers
- version
- Versus
- via
- Voir
- viral
- virus
- virus
- vivo
- volumes
- était
- Washington
- Eau
- we
- semaine
- Semaines
- poids
- WELL
- Wells
- ont été
- qui
- la totalité
- répandu
- comprenant
- dans les
- world
- X
- années
- Rendement
- rendements
- Zen
- zéphyrnet