انباکو نوشی
All incubation processes were carried out in a ThermoMixer C (Eppendorf) except for Supplementary Figs. 1 اور 9 (Dry Bath FB15103 incubator, Fisher Scientific) and for Fig. 1c اور اضافی انجیر 3-6 (QuantStudio5, Applied Biosystems by Thermo Fisher Scientific).
Isothermal self-assembly of 2D DNA origamis
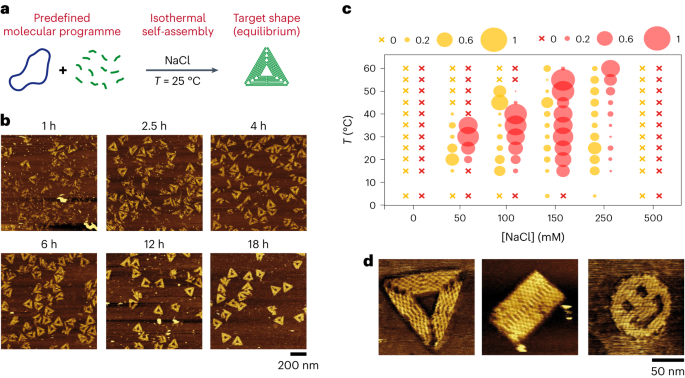
تصویر دیکھیں۔ 1 اور اضافی انجیر 1 9 اور 11 14. We used the staple cocktail without any thermal pretreatment and directly mixed it with the desired buffer prior to brief vortexing and addition of the M13 template (1 nM) to the solution and gentle up-and-down mixing with a pipette. The solution was left to incubate, without further mixing, at a fixed temperature for the desired amount of time.
Thermal annealing of DNA origamis in TANa buffer
See Supplementary Fig. 10. We assembled the M13 template (1 nM) with a mixture of staples (40 nM each staple) in TANa supplemented with 100 mM of NaCl. The sample was incubated for 10 min at 90 °C and then subjected to a thermal ramp in a peqSTAR 2X thermocycler (Peqlab) from 70 °C to 20 °C at a rate of −1 °C per 10 min.
Purification by PEG precipitation
See Supplementary Figs. 13 اور 14. DNA origamis obtained by isothermal assembly in TANa buffer ([NaCl] = 100 mM) at 25 °C were purified from their staple strands by PEG precipitation. The method was inspired by the protocol introduced in a previous report35. The DNA origamis were diluted three times with a solution of PEG 8000 and NaCl to reach final concentrations of 4% w/v and 500 mM, respectively. After gentle mixing, the solution was left to incubate for 15 min at room temperature and centrifuged at 15,000g for 15 min. The supernatant was removed and the origamis were resuspended to their initial volume in TANa buffer ([NaCl] = 100 mM). If necessary, the process was repeated for a second consecutive purification.
Gel electrophoresis of purified origamis
See Supplementary Fig. 13. We prepared 50 ml of agarose (type I low EEO, Sigma Aldrich) gel at 1.5% containing 4 μl of GR-Green 10,000× (Excellgen) in TBE 1× buffer. After the gel had cooled down, we introduced, in each well, 18 µl of 100 bp DNA Ladder (New England Biolabs) or 18 µl of sample supplemented with 1× of DNA loading dye SDS solution (Thermo Scientific). The migration was performed at 100 V for 1 h in a 7 cm electrophoresis cell filled with TBE 1× buffer.
Isothermal stepwise assembly
See Supplementary Fig. 24. The staples of the triangle were assembled into three separate lots, each coding for the top corner, the intermediate part and the opposite edge. One lot (40 nM each staple) was mixed with M13 (1 nM) in TANa buffer ([NaCl] = 100 mM) and the system was left to incubate at 25 °C without further mixing. Every 24 h, we removed the volume necessary for AFM imaging, added one lot coding for an additional part of the triangle (40 nM each staple) and let the system incubate at 25 °C in TANa buffer ([NaCl] = 100 mM). We performed two different ways of stepwise assembly, from the corner to the opposite side and from one side to the opposite corner.
Isothermal preparation of streptavidin-modified triangles
تصویر دیکھیں۔ 2a اور اضافی انجیر 15 اور 16. In the same tube, we mixed 1 nM of M13, 40 nM of each of the staples including biotinylated ones, and 2 µM of streptavidin in TANa buffer supplemented with 100 mM of NaCl. The sample was left to incubate at 25 °C without further mixing for 24 h.
Isothermal preparation of SST R4 rectangles
تصویر دیکھیں۔ 2b. We mixed all strands of the R4 rectangle in the buffer to a final concentration of 100 nM in each strand in TANa supplemented with 100 mM NaCl. The sample was left to incubate at 25 °C without further mixing for 24 h.
Gel electrophoresis of SST R4 rectangles
تصویر دیکھیں۔ 2b. A 1.5% agarose gel (type I low EEO, Sigma Aldrich) was prepared in TBE 0.5× buffer supplemented with 11 mM MgCl2 and GB green DNA stain. Gel electrophoresis was performed in an ice-water bath for 2 h at 100 V of voltage using a 1 kb DNA ladder. For purification, the target band of the gel was cut into small pieces and put into a tube with a spin column, and column was subjected to centrifugation at 5,000g for 10 min. For AFM imaging, eluted sample was directly adsorbed on a mica plate for 10 min inside the atmosphere-controlled chamber. The sample was then rinsed with 1 ml of in 0.5× TBE + 11 mM MgCl2 and observed using AFM in 0.5× TBE + 11 mM MgCl2.
Isothermal preparation of DNA nanogrids
تصویر دیکھیں۔ 2c اور ضمنی تصویر۔ 17. We mixed the nine oligonucleotides (1 µM of each nucleotide) in TANa buffer supplemented with 100 mM or 150 mM of NaCl. The sample was left to incubate at 25 °C without further mixing for 24 h.
Thermal annealing of 3D origamis
تصویر دیکھیں۔ 3a اور ضمنی تصویر۔ 18. The scaffold (7,560 nt M13 for Tb, 8,064 nt M13 for T1) and the staple mix (10× excess in each staple) were mixed in buffer containing 5 mM Tris–HCl, pH 8.0, 1 mM EDTA and 18 mM MgCl2. The mixture was heated to 65 °C for 15 min to denature all DNA strands prior to being slowly cooled down in a gradient from 60 °C to 40 °C, over 41 h to anneal and assemble the 3D origami nanostructures.
Negative-stain TEM
تصویر دیکھیں۔ 3 اور ضمنی تصویر۔ 18. For TEM characterization, DNA nanostructures were first purified from 1% agarose (0.5× TBE, 45 mM Tris–borate, 1 mM EDTA, pH 8.3) supplemented with 11 mM MgCl2 and 0.5 mg ml1- Sybr SAFE. Samples were migrated on the gel for 3 h with a running buffer of 0.5× TBE, 11 mM MgCl2 at 2.85 V cm1- at room temperature. Bands corresponding to self-assembled structures were excised and transferred to a DNA gel-extraction spin column (Merck) and centrifuged at 5,000g for 5 min at 4 °C. Purified origamis were then deposited by adsorption onto glow-discharged carbon-coated grid (Quantifoil Micro Tools), stained for 60 s with a 2% (w/v) aqueous uranyl acetate (Merck) solution and then dried with ashless filter paper (VWR). TEM observations were carried out on a JEM-1400 Flash Tungsten microscope working at 120 kV, equipped with a Gatan OneView camera.
Isothermal competition
See Supplementary Fig. 20. We briefly vortexed the mixtures of two staple sets coding for triangles and rectangles (40 nM final concentration for each staple) with the buffer (TANa supplemented with 100 mM NaCl) prior to adding M13 to the mixture (1 nM final concentration) and gentle mixing with a pipette. The sample was left to incubate at 25 °C without further mixing.
AFM observation of DNA nanostructures in liquid
Environmental high-resolution AFM observations in sample buffer were used for all AFM data and images shown in this article. Except for Fig. 4 (see the specific protocol below), DNA nanostructures obtained in TANa buffer (DNA origamis with or without protein modification, SST R4 rectangles, DNA nanogrids) were adsorbed on freshly cleaved 10-mm-diameter mica discs (Nano-Tec V-1 grade Muscovite, Micro to Nano Innovative Microscopy Supplies) previously glued to a metal disc and treated with 20 µl of a spermine tetrachloride solution (0.1 M in MilliQ water) for 10 min and washed abundantly, first with MilliQ water and then with the TANa buffer. For sample adsorption, 15–20 µl of sample was deposited on the freshly spermine-treated mica and left to adsorb for 10 min, except for isothermal transformation experiments (Fig. 5 اور اضافی انجیر 21-23) where the time was increased to 20 min due to the lower concentration of origamis. The mica plate was then gently rinsed with 200 µl of the buffer to remove the excess of staples and non-adsorbed objects. To prevent the drying of the sample during mica manipulation, we left a thin layer of buffer on the top of the adsorbed sample and kept it at room temperature in an atmosphere-controlled chamber (a sealed container containing a piece of Kimtech wipe wetted with MilliQ water). The same protocol was done with samples containing magnesium (Supplementary Fig. 1, TAEMg and TAMg buffers) except that the samples were directly adsorbed for 5 min on the freshly cleaved mica without any treatment. The samples were observed with a Cypher ES atomic force microscope (Oxford Instruments) in tapping mode with 17–45 kHz resonance frequency in liquid and a 0.09 N m1- force constant tip (BL-AC40TS, Olympus), using the blueDrive photothermal excitation mode. Raw images were subjected to polynomial background subtraction, plane-level correction, row alignment using various methods and horizontal scar correction in Gwyddion.
Real-time imaging of the Λ → Δ isothermal evolution on a lipid bilayer surface
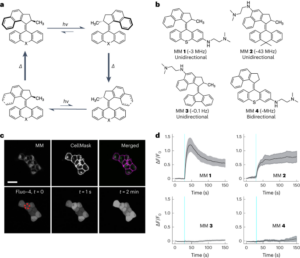
تصویر دیکھیں۔ 4, Supplementary Text 3 اور سپلیمنٹری موویز 1 4. Supported lipid bilayers (SLBs) were obtained from DOPC liposomes via our previously described method37. Vesicles were prepared from a chloroform stock of DOPC. After evaporation of the chloroform under a stream of nitrogen gas, the lipids were rehydrated in MilliQ water to reach a final lipid concentration of 2 mg ml1-. The lipid mixture was then vortexed and sonicated for 60 min to produce small unilamellar vesicles. To prevent the drying of the bilayers, the following steps were carried out in an atmosphere-controlled chamber. SLBs were formed by depositing 2 ml of the vesicle solution onto freshly cleaved mica discs (previously stuck on a magnetic metallic plate with glue), followed by 2 µl of TAEMg (Tris–acetate 1×, [EDTA] = 1 mM, [MgCl2] = 12.5 mM). After 30 min of adsorption, the sample was rinsed with 2 µl of TAEMg buffer to remove unadsorbed liposomes and this adsorption process was repeated a second time to ensure optimal coverage of the mica surface by the bilayer. At the end of the adsorption process, the sample was rinsed with 5 µl of TAENa (Tris–acetate 1×, [EDTA] = 1 mM, [NaCl] = 100 mM) to ensure that there will be no remaining free Mg2+ ions on the sample, which would prevent isothermal folding of the origamis.
Cholesterol-modified Λ-shaped origamis (Supplementary Fig. 19) were prepared by mixing a solution of 10 nM of M13 in the TAENa buffer with 20 nM of each of the staples and annealing by reducing the temperature from 70 °C to 4 °C at a rate of −0.1 °C min1-. The resulting origami solution was used without further purification. Then, 2 µl of the cholesterol-modified Λ origamis solution was deposited onto the preformed SLB, followed by 2 µl of TAENa buffer. The sample was incubated for 60 min at room temperature in the atmosphere-controlled chamber, and the surface was then directly imaged at room temperature (T = 26 °C) by AFM in 20 µl of TAENa buffer without surface rinsing. After selecting a position containing a sufficient number of Λ origamis adsorbed on the SLB, 8 µl of the A-side staples in TAENa were added onto the sample without moving it or removing it from the AFM stage. The solution overhanging the sample was then gently mixed by slowly moving the AFM probe up and down several times to accelerate the diffusion on the A-side staples towards the mica surface. The same position was then scanned on average every 3 min for 223 min. The image resolution was 512 × 512 from t = 0 سے t = 41 min and was then changed to 640 × 640. The z scale of the images was 0–10 nm from t = 0 سے t = 47 min and 0–7 nm afterwards. Because of the evaporation during the imaging process, supplementary buffer (8 µl) was added at t = 63 min, t = 144 min and t = 161 min, and 8 µl of the A-side staples were furthermore added after t = 170 min.
AFM images were obtained in TAENa at room temperature using a Brücker Fast Scan atomic force microscope in tapping mode. All AFM experiments were performed using Olympus probes. Images obtained by this protocol are displayed in Fig. 4 اور سپلیمنٹری موویز 1-4.
Isothermal morphological transformation
تصویر دیکھیں۔ 5 اور اضافی انجیر 21 23. DNA rectangle origamis, without or with shortened staples, were first prepared by thermal annealing: after assembly of 1 nM of the M13 template with a mixture of staples (40 nM each) in TANa buffer (with 100 mM or 150 mM NaCl), the sample was incubated for 10 min at 90 °C and then subjected to a thermal ramp in a peqSTAR 2X thermocycler (Peqlab) from 70 °C to 20 °C at a rate of −0.1 °C per 2 min. Triangle staples were then mixed directly to the sample, without any purification (rectangle staples are kept in the system), with a pipette to a desired concentration (10 or 100 nM of each staple) with a final concentration of 0.25 nM in [M13] and 10 nM in each rectangle staple. The resulting sample was left to incubate at 25 °C or 30 °C without further mixing.
شماریات اور تولیدی صلاحیت
Except for the experiment displayed in Fig. 4, which was only performed once due to the set-up complexity, all other investigations were replicated several times to ensure reproducibility. All the AFM image analyses were performed on a large number n of individual origamis taken from different images, at different positions of samples obtained in the same conditions. All detected origamis were analysed. No origami, properly folded or not, was excluded from these analyses. The number n of analysed objects in each condition shown in the different figures is displayed in Supplementary Tables 1-5. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
- SEO سے چلنے والا مواد اور PR کی تقسیم۔ آج ہی بڑھا دیں۔
- پلیٹو ڈیٹا ڈاٹ نیٹ ورک ورٹیکل جنریٹو اے آئی۔ اپنے آپ کو بااختیار بنائیں۔ یہاں تک رسائی حاصل کریں۔
- پلیٹوآئ اسٹریم۔ ویب 3 انٹیلی جنس۔ علم میں اضافہ۔ یہاں تک رسائی حاصل کریں۔
- پلیٹو ای ایس جی۔ آٹوموٹو / ای وی، کاربن، کلین ٹیک، توانائی ، ماحولیات، شمسی، ویسٹ مینجمنٹ یہاں تک رسائی حاصل کریں۔
- بلاک آفسیٹس۔ ماحولیاتی آفسیٹ ملکیت کو جدید بنانا۔ یہاں تک رسائی حاصل کریں۔
- ماخذ: https://www.nature.com/articles/s41565-023-01468-2
- : ہے
- : نہیں
- :کہاں
- $UP
- 1
- 10
- 100
- 11
- 12
- 15٪
- 2%
- 20
- 200
- 2014
- 2015
- 24
- 25
- 26
- 2D
- 30
- 3d
- 40
- 50
- 500
- 60
- 65
- 7
- 70
- 8
- 90
- a
- رفتار کو تیز تر
- شامل کیا
- انہوں نے مزید کہا
- اس کے علاوہ
- ایڈیشنل
- کے بعد
- بعد
- تمام
- تین ہلاک
- رقم
- an
- لنگر
- اور
- کوئی بھی
- اطلاقی
- کیا
- مضمون
- جمع
- اسمبلی
- تشخیص
- At
- اوسط
- پس منظر
- بینڈ
- BE
- کیونکہ
- کیا جا رہا ہے
- نیچے
- BP
- مختصر
- بفر
- by
- کیمرہ
- کیا ہوا
- سیل
- چیمبر
- تبدیل کر دیا گیا
- کلک کریں
- کاک
- کوڈنگ
- کالم
- پیچیدگی
- دھیان
- شرط
- حالات
- مسلسل
- مسلسل
- کنٹینر
- کونے
- اسی کے مطابق
- کوریج
- کٹ
- سائپر
- اعداد و شمار
- گھنے
- جمع
- بیان کیا
- مطلوبہ
- پتہ چلا
- مختلف
- براڈ کاسٹننگ
- براہ راست
- ظاہر
- ڈی این اے
- کیا
- نیچے
- خشک
- دو
- کے دوران
- e
- ہر ایک
- ed
- ایج
- آخر
- انگلینڈ
- کو یقینی بنانے کے
- لیس
- Ether (ETH)
- ہر کوئی
- ارتقاء
- اس کے علاوہ
- اضافی
- خارج کر دیا گیا
- تجربہ
- تجربات
- فاسٹ
- انجیر
- اعداد و شمار
- اعداد و شمار
- بھرے
- فلٹر
- فائنل
- پہلا
- مقرر
- فلیش
- پیچھے پیچھے
- کے بعد
- کے لئے
- مجبور
- تشکیل
- مفت
- فرکوےنسی
- سے
- مزید
- مزید برآں
- گیس
- نرم
- گریڈ
- سبز
- گرڈ
- تھا
- بهترین ریزولوشن
- افقی
- HTTPS
- i
- if
- تصویر
- تصاویر
- امیجنگ
- in
- سمیت
- اضافہ
- انکیوبیٹڈ
- انکیوبیشن
- انکیوبیٹر
- انفرادی
- ابتدائی
- جدید
- کے اندر
- متاثر
- آلات
- انٹرمیڈیٹ
- میں
- متعارف
- تحقیقات
- تحقیقاتی
- IT
- رکھی
- سیڑھی
- بڑے
- پرت
- چھوڑ دیا
- LINK
- مائع
- لوڈ کر رہا ہے
- بہت
- لو
- کم
- ہیرا پھیری
- مارٹن
- مواد
- مرک
- دھات
- طریقہ
- طریقوں
- ایم سی اے
- مائکرو.
- خوردبین
- خوردبین
- منتقل
- منتقلی
- منٹ
- اختلاط
- مخلوط
- مخلوط
- مرکب
- ML
- موڈ
- فلم
- منتقل
- نینو
- نےنو
- فطرت، قدرت
- ضروری
- نئی
- نہیں
- تعداد
- اشیاء
- مشاہدہ
- حاصل کی
- of
- اولمپکس
- on
- ایک بار
- ایک
- والوں
- صرف
- اس کے برعکس
- زیادہ سے زیادہ
- or
- دیگر
- ہمارے
- باہر
- نتائج
- پر
- آکسفورڈ
- کاغذ.
- حصہ
- پت
- فی
- کارکردگی
- ٹکڑا
- ٹکڑے ٹکڑے
- پلاٹا
- افلاطون ڈیٹا انٹیلی جنس
- پلیٹو ڈیٹا
- پوزیشن
- پوزیشنوں
- تیاری
- تیار
- کی روک تھام
- پچھلا
- پہلے
- پہلے
- تحقیقات
- عمل
- عمل
- پیدا
- مناسب طریقے سے
- پروٹین
- پروٹوکول
- ڈال
- ریمپ
- بے ترتیب
- شرح
- خام
- تک پہنچنے
- کو کم کرنے
- باقی
- ہٹا
- ہٹا دیا گیا
- کو ہٹانے کے
- بار بار
- نقل تیار
- قرارداد
- گونج
- بالترتیب
- نتیجے
- کمرہ
- ROW
- چل رہا ہے
- s
- محفوظ
- اسی
- توسیع پذیر
- پیمانے
- اسکین
- سائنسی
- دوسری
- دیکھنا
- منتخب
- علیحدہ
- سیٹ
- کئی
- قصر
- دکھایا گیا
- کی طرف
- سگما
- سائز
- آہستہ آہستہ
- چھوٹے
- حل
- حل
- مخصوص
- سپن
- اسٹیج
- شماریات
- مراحل
- اسٹاک
- اسٹریڈز
- سٹریم
- کافی
- تائید
- سطح
- کے نظام
- T
- T1
- لیا
- ٹیپ
- ہدف
- سانچے
- کہ
- ۔
- ان
- تو
- وہاں.
- تھرمل
- یہ
- اس
- تین
- وقت
- اوقات
- ٹپ
- کرنے کے لئے
- اوزار
- سب سے اوپر
- کی طرف
- منتقل
- تبدیلی
- علاج کیا
- علاج
- دو
- قسم
- کے تحت
- استعمال کیا جاتا ہے
- کا استعمال کرتے ہوئے
- مختلف
- کی طرف سے
- وولٹیج
- حجم
- تھا
- پانی
- طریقوں
- we
- اچھا ہے
- تھے
- جس
- گے
- مسح
- ساتھ
- بغیر
- کام کر
- گا
- زیفیرنیٹ