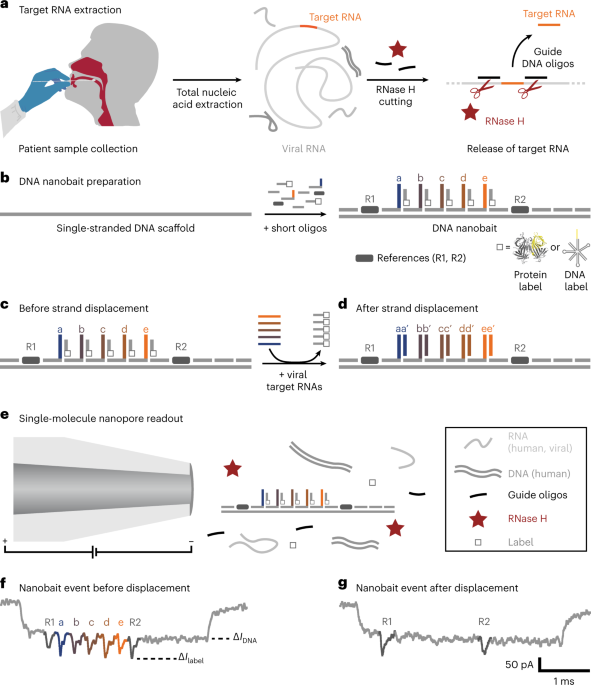
Збір зразків пацієнта
Зразки пацієнтів збирали шляхом взяття мазка із задньої стінки горла (мазка з ротоглотки) пацієнтів, як описано раніше26. Зразки були відібрані у пацієнтів із клінічною картиною, схожою на COVID-19, і перевірені за допомогою qRT-PCR після екстракції нуклеїнової кислоти. Коротко кажучи, після збору тампони поміщали в мічену пробірку для зразків, що містить буфер для лізису (4 М гуанідинтіоціанат, 25 мМ Трис-HCl, 0.5% β-меркаптоетанол і РНК MS2 (200 нг мкл).-1; Рош)). Пробірку обережно перемішували, щоб забезпечити рівномірний розподіл буфера для лізису. Заходи безпеки були описані раніше та виконані в сертифікованій лабораторії CL226.
Екстракція нуклеїнової кислоти
Загальну нуклеїнову кислоту екстрагували за допомогою систем на основі спінової колонки та стандартизованого тестування qRT-PCR26. Внутрішній контроль посилення (MS2 (~6 × 104 БФУ мл-1) на 10 мл буфера для лізису) додавали в буфер для лізису (25 мкл на 10 мл буфера для лізису). Зразок елюювався 100 мкл води, вільної від нуклеаз (nfH2О; Invitrogen) і залишають на 1 хвилину перед центрифугуванням протягом 1 хвилини при 21,130 ×g (15,000 80 об/хв) у настільній мікроцентрифузі. Елюйовані зразки безпосередньо піддавали QRT-PCR. Решту екстрактів нуклеїнових кислот зберігали при -XNUMX °C і далі використовували для зондування наноприманки-нанопори.
qRT-PCR для SARS-CoV-2
Виявлення SARS-CoV-2 було виконано, як описано раніше26. На реакцію основна суміш містила 12.5 мкл одноетапної реакційної суміші 2× Luna Universal Probe, 0.5 мкл 20 мкМ прямого праймера Wu (5′-ATGGGTTGGGATTATCCTAAATGTGA-3′), 0.5 мкл 20 мкМ зворотного праймера Wu (5′ -GCAGTTGTGGCATCTCCTGATGAG-3′), 0.3 мкл 10 мкМ MGB Probe 3 флуоресцеїну (5′-ATGCTTAGAATTATGGCCTCAC-3′), 0.5 мкл 10 мкМ прямого праймера внутрішнього контролю для РНК MS2, 0.5 мкл 10 мкМ внутрішнього контрольного зворотного праймера для РНК MS2, 0.3 мкл 10 мкМ внутрішнього зонда (MS2 ROX), 1 мкл суміші ферментів Luna WarmStart RT і 3.9 мкл nfH2О. Потім 20 мкл основної суміші розподіляли в кожну лунку 96-лункового планшета, а потім об’єднували з 5 мкл кожного екстракту. Контроль внутрішньої екстракції та ампліфікації MS2, який пройшов повний протокол екстракції, був включений як негативний контроль екстракції принаймні у дві лунки планшета для ПЛР-ПЛР. Для визначення потенційного забруднення в процесі qRT-PCR, 5 мкл nfH2O був включений як негативний контроль QRT-PCR. Потім 5 мкл матричної плазміди SARS-CoV-2 з додаванням додали в одну лунку як позитивний контроль qRT-PCR. Після додавання 5 мкл кожного зразка до призначеної лунки планшет закривали оптично прозорою пластиковою плівкою. Планшет центрифугували протягом 1 хв при 2,000×g (1,000 об/хв) при 4 °C, а потім вставили в апарат qRT-PCR (QuantStudio, Thermo Fisher Scientific), і цикл параметризували. Були отримані сигнали для флуоресцеїну (FAM) і карбоксиродаміну (ROX). ROX використовувався для виявлення внутрішнього контролю MS2, а флуоресцеїн використовувався для виявлення РНК SARS-CoV-2. Аналіз проводили протягом 2 хвилин при 25 °C, 15 хвилин при 50 °C (для зворотної транскриптази), 2 хвилини при 90 °C, перед тим, як 45 циклів 95 °C протягом 3 секунд, потім 60 °C протягом 30 секунд . Результати визначали за підтвердженням правильності позитивних контролів (ампліфікація плазміди), контролів екстракції та ампліфікації всіх зразків (канал ROX), відсутності ампліфікації в негативних контролях і постійних середніх значень контролів. Позитивність SARS-CoV-2 була підтверджена ампліфікацією у флуоресцеїновому каналі за допомогою відповідної сигмоїдальної кривої зі значенням CT ≤36. Значення КТ для зонда MS2 і MGB 3 підтримувалися для відстеження якості та відтворюваності аналізу44.
Програмоване різання РНКази H для наноприманки
Для зондування нанопор контрольні зразки РНК SARS-CoV-2, екстракти нуклеїнових кислот (зразки пацієнтів) або РНК вірусу MS2 використовувалися для виявлення за допомогою наноприманки. Спочатку ми змішали направляючі олігонуклеотиди зі зразком і нагріли його до 70 °C протягом 5 хв. РНКазу H (5,000 одиниць на мл; NEB) додавали, змішували та нагрівали протягом 20 хвилин при 37 °C, щоб дозволити ферменту розрізати РНК у гібриді ДНК: РНК, який ефективно вивільняє цільову РНК. РНКазу H термічно інактивували шляхом інкубації при 65 °C протягом 10 хв. Керівні олігонуклеотиди перевірені на відсутність внутрішньомолекулярних структур, гомо- або гетеродимерів за допомогою програмного забезпечення NUPACK45. Для вимірювання з відсутньою мішенню використовувався той самий протокол, що включає направляючі олігонуклеотиди. Контрольні вимірювання не показують зміщення, і, отже, ми можемо виключити будь-яке суттєве перехресне зв’язування з направляючими олігонуклеотидами.
Властивості вірусної цільової послідовності для наноприманки
Довжина мішені, довжина опори та вміст GC були обрані для забезпечення оптимальної гібридизації21. Для дизайну ДНК-наноприманок цільові послідовності були вибрані так, щоб вони знаходились у збережених областях вірусного геному та мали 40–60% вміст GC для формування стабільного дуплексу 20 bp. Довжина опори була вибрана такою, щоб вона становила 6 нт і мала 40–60% вміст GC. Ми перевірили всі послідовності на потенційні небажані високостабільні внутрішньомолекулярні взаємодії або гомодимери за допомогою програмного забезпечення NUPACK (веб-додаток 2020)45. Потім ми провели перевірку перехресної реактивності між кількома сайтами, задіяними в кожному експерименті45.
Підготовка квітки ДНК для наноприманки
Ми розробили квітку ДНК як ще одну мітку для виявлення РНК SARS-CoV-2 у зразках пацієнтів. Три квітки ДНК, специфічні для кожної мішені SARS-CoV-2 (семисторонні з’єднання, 7WJa, 7WJb і 7WJc), були підготовлені окремо. Беручи 7WJc як приклад, 4 мкМ ланцюг ДНК J1, J2, J3 і J4c (додаткова таблиця 1) змішували разом у буфері ТМ (10 мМ Tris–HCl, 10 мМ MgCl2, pH 8.0) і нагрівають до 90 °C протягом 5 хвилин, потім охолоджують до 65 °C протягом 15 хвилин, 45 °C протягом 15 хвилин, 37 °C протягом 20 хвилин, 25 °C протягом 20 хвилин і, нарешті, до 4 °C. C протягом 20 хв. Нитка J4c була замінена на J4b для отримання 7WJb. Для 7WJa, щоб уникнути самозгортання в місці 43 на наноприманці, J1, J2, J3 J4a і C43 були змішані разом перед відпалом.
Самозбірка наноприманки ДНК
ДНК-наноприманку збирали шляхом змішування лінеаризованої одноланцюгової ДНК M13 (M13mp18, 7,249 нт, Guild Biosciences, 100 нМ) з короткими комплементарними олігонуклеотидами.12 (деякі з яких містили еталонні структури та ланцюги захоплення) і шляхом додавання частково комплементарних ланцюгів, які були 3'-біотинільованими для реакції зміщення ланцюга, опосередкованої опорою. Лінеаризована ДНК M13 (довжиною 7,228 нт) була доповнена олігонуклеотидами, таким чином створивши подвійну ланцюгову наноприманку з розрізами з двома кінцевими чотирма дезокситимідиновими виступами, які запобігають мультимеризації12. Суміш містила 20 нМ лінеаризованої ДНК М13, 60 нМ олігонуклеотидів (трикратний надлишок до ДНК М13), 3′-біотинільовані нитки в концентрації 180 нМ, 10 мМ MgCl.2 і 1× TE (10 мМ Tris–HCl, 1 мМ EDTA, pH 8.0). Його змішували піпетуванням і центрифугували перед нагріванням до 70 °C протягом 30 секунд і охолоджували протягом 45 хвилин до температури навколишнього середовища. Надлишок олігонуклеотидів видаляли за допомогою відцентрових фільтрів Amicon Ultra 0.5 мл з відсіканням 100 кДа з промивним буфером (10.0 мМ Tris–HCl pH 8.0, 0.5 мМ MgCl2). Якщо як мітку використовували квіти ДНК, частково комплементарні ланцюги, які несуть її, інкубували в 10 мМ MgCl2 протягом 2 годин при температурі навколишнього середовища, а потім проводили фільтрацію Amicon, як описано вище. Асиметрія дизайну наноприманки дозволяє однозначно ідентифікувати сайти зв’язування. Наноприманку зберігали до використання для подальших експериментів при 4–10 °C у 0.5 мМ MgCl2, 10.0 мМ Трис–HCl, pH 8.0. Конструкцію наноприманки перевіряли зчитуванням нанопор перед кожним вимірюванням.
Зчитування нанопор ДНК наноприманки
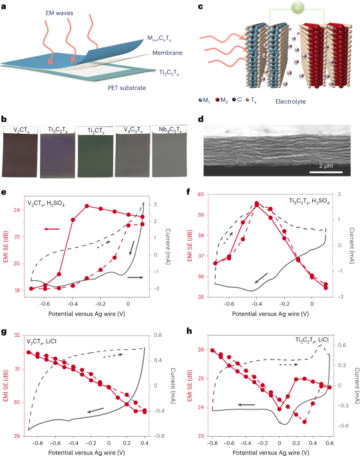
Наноприманку змішували зі зразком (екстракт нуклеїнової кислоти або очищені вірусні мішені у десятикратному надлишку) у 10 мМ MgCl2 і 100 мМ NaCl. Суміш (5 мкл) інкубували при кімнатній температурі (~10 хв) до підготовки до вимірювання нанопор. Різниця в складі цільової послідовності та її фізичних характеристиках може призвести до мінливості гібридизації та, отже, ефективності заміщення сайтів зондування21. Ми використовували htRNA (100 нг мкл-1; Invitrogen) як фон, де зазначено, щоб показати відсутність неспецифічних сигналів, індукованих природними РНК людини. Для вимірювання нанопор зразок розбавляли до <0.5 нМ наноприманки (для очищених вірусних мішеней) або 4.7 мкл зразка пацієнта, розрізаного РНКазою-Н, змішували з 0.3 мкл моновалентного стрептавідину (SAe1D3)18 (1 мкМ), 5 мкл LiCl (4.0 М) і 5.0 мкл LiCl (8.0 М). Ми виготовили нанопори розміром 14 ± 3 нм (середнє значення ± стандартне відхилення).12 з використанням капілярів із кварцового скла із зовнішнім діаметром 0.5 мм і внутрішнім діаметром 0.2 мм (Sutter Instrument) за допомогою лазерного пулера P-2000 (Sutter Instrument). Суміш піпетували в нанопористий полідиметилсилоксановий чіп, і всі вимірювання проводили при постійній напрузі 600 мВ. Деталі вимірювання нанопор наведено в додатковій таблиці 30.
Аналіз даних про нанопори в реальному часі
Аналіз даних про нанопори детально пояснюється в Додатковому розділі 14. Коротко кажучи, події наноприманки були відфільтровані з необроблених слідів іонного струму, а потім була визначена область виявлення та отримана інформація про присутність спайку в кожному конкретному місці. Нанесену на графік ефективність заміщення розраховували як ефективність заміщення для вимірювання, відраховану від нецільового контролю для кожного місця (50 подій наноприманки для кожного з трьох записів нанопор), якщо не зазначено інше:
$$begin{array}{l}{mathrm{Displacement}},{mathrm{efficiency}} =frac{1}{3}mathop {sum}limits_{n = 1}^3 left{ {1 -frac{1 }{{50}}mathop {sum}обмеження_{n = 1}^{50} {left[ {fleft( n right) = left( {frac{{1,,mathrm{peak}}}{{0,, {mathrm{no}},{mathrm{peak}}}}} right)} right]_{{{{mathrm{target}}}}}} } right}\ – frac{1}{3}mathop {sum }limits_{n = 1}^3 {left{ {1 – frac{1}{{50}}mathop {sum }limits_{n = 1}^{50} left[ {fleft( n right) = left( { frac{{1,,mathrm{peak}}}{{0,,mathrm{no}},{mathrm{peak}}}} right)} right]_{{{{mathrm{no}}}},{ {{mathrm{target}}}}}} right}} end{array}.$$
Ми перевірили, що згорточна нейронна мережа QuipuNet27 був здатний аналізувати дані про нанопори в режимі реального часу за описаною процедурою. Раніше ми продемонстрували, що приблизно з десятьма подіями ми досягаємо 99% впевненості в позитивному виявленні розроблених нами структур ДНК46.
АСМ зображення
AFM (Nanosurf Mobile S) зображення наноприманок проводили на повітрі в безконтактному режимі. Структури наноприманки розводили до 1 нг мкл-1 в 1 мМ MgCl2 і 10 мкл додавали до свіжорозколотої слюди, інкубували протягом 1 хв, промивали відфільтрованою водою Milli-Q і потім сушили феном з азотом. Перед скануванням слюдяна пластина була прикріплена до столика зразка АСМ за допомогою двосторонньої клейкої стрічки. Візуалізацію та аналіз зображень проводили за допомогою Gwyddion (версія 2.60).
Статистичний аналіз
Для всіх вимірювань було розраховано 99.9% довірчі інтервали для ефективності витіснення. Статистичну значущість між двома сайтами без і з ціллю перевіряли за допомогою двостороннього Стьюдента t-тест.
Підсумок звітності
Більш детальна інформація про розробку досліджень доступна в Короткий звіт про портфоліо природи пов'язана з цією статтею.
- Розповсюдження контенту та PR на основі SEO. Отримайте посилення сьогодні.
- Платоблокчейн. Web3 Metaverse Intelligence. Розширені знання. Доступ тут.
- джерело: https://www.nature.com/articles/s41565-022-01287-x
- 000
- 1
- 10
- 100
- 11
- 110
- 2014
- 2016
- 2017
- 2018
- 2020
- 2021
- 7
- 70
- 9
- a
- вище
- відсутнім
- академічний
- придбаний
- доданий
- після
- AIR
- ВСІ
- дозволяє
- Ambient
- аналіз
- Якір
- та
- Інший
- додаток
- підхід
- відповідний
- навколо
- стаття
- зібраний
- доступний
- назад
- фон
- перед тим
- Дзвін
- між
- обов'язковий
- підривати
- BP
- коротко
- буфера
- розрахований
- здатний
- захоплення
- нести
- Сертифікований
- Канал
- характеристика
- перевірка
- Чень
- чіп
- ясно
- Клінічний
- збір
- комбінований
- взаємодоповнюючі
- концентрація
- довіра
- Підтверджено
- послідовний
- постійна
- зміст
- контроль
- управління
- згорткова нейронна мережа
- створення
- Поточний
- крива
- Вирізати
- різання
- циклів
- дані
- аналіз даних
- продемонстрований
- описаний
- дизайн
- призначений
- конструкцій
- деталь
- деталі
- Виявлення
- Визначати
- певний
- відхилення
- різниця
- в цифровому вигляді
- безпосередньо
- розподіл
- ДНК
- вниз
- кожен
- фактично
- Ефективність
- ефективність
- Машинобудування
- забезпечувати
- помилка
- Ефір (ETH)
- Навіть
- Події
- приклад
- пояснені
- витяг
- видобуток
- Виписки
- Фільтри
- в кінці кінців
- Перший
- квітка
- потім
- після
- форма
- Вперед
- від
- Повний
- далі
- геном
- скло
- керівництво
- дуже
- HTTPS
- людина
- гібрид
- Ідентифікація
- зображення
- Зображеннями
- реалізація
- in
- включені
- У тому числі
- інкубований
- ІНКУБАЦІЯ
- інформація
- інструмент
- Взаємодії
- внутрішній
- Іонний
- IT
- КДА
- етикетка
- вести
- довжина
- LINK
- пов'язаний
- Довго
- Місяць
- машина
- майстер
- матеріал
- вимірювання
- Слюда
- може бути
- мінімальний
- змішаний
- Змішування
- суміш
- ML
- Mobile
- режим
- MOL
- множинний
- нано
- нанопор
- рідний
- природа
- негативний
- мережу
- Нейронний
- нейронної мережі
- відкрити
- оптимальний
- інакше
- спаровування
- пацієнт
- pacientes
- продуктивність
- фізичний
- картина
- пластик
- plato
- Інформація про дані Платона
- PlatoData
- портфель
- позитивний
- Позитивність
- потенціал
- Готувати
- підготовлений
- наявність
- запобігати
- раніше
- Праймер для вій
- зонд
- процес
- властивості
- Білок
- протокол
- якість
- Сировина
- досягати
- реакція
- реального часу
- регіон
- райони
- Релізи
- решті
- Вилучено
- Звітність
- дослідження
- результати
- зворотний
- РНК
- рок
- Кімната
- rt
- Правила
- прогін
- Безпека
- то ж
- ТОРС-коронавірус-2
- сканування
- розділ
- обраний
- Послідовність
- Короткий
- Показувати
- показаний
- сигнали
- значення
- одночасний
- один
- сайт
- сайти
- невеликий
- Софтвер
- деякі
- конкретний
- крутився
- стабільний
- Стажування
- стояти
- standard
- заявив,
- статистичний
- заходи
- зберігати
- Пасма
- Згодом
- істотний
- Systems
- таблиця
- взяття
- Мета
- цілі
- шаблон
- десять
- Команда
- тим самим
- три
- times
- TM
- до
- разом
- Усього:
- трек
- Ультра
- при
- одиниць
- Universal
- підтверджено
- значення
- Цінності
- перевірено
- версія
- через
- віруси
- візуалізації
- Напруга
- W
- вода
- Web
- Веб-додаток
- Wells
- який
- без
- wu
- зефірнет