Food and Drug Administration (FDA) spiller en sentral rolle i å ivareta folkehelsen gjennom regulering av medisinsk utstyr, og sikrer at de er både trygge og effektive for den tiltenkte bruken. I moderne tid har fremveksten av tilkoblede enheter brakt cybersikkerhet i forkant av FDA-bekymringer; i denne sammenhengen er det viktig å definere spesifikke krav for definisjonen av cybersikkerhetstrusselmodellen. Etter hvert som medisinsk utstyr blir stadig mer integrert med teknologi, er behovet for robuste cybersikkerhetstiltak for å beskytte pasientinformasjon og sikre funksjonaliteten til disse enhetene mer kritisk enn noen gang. Overholdelse av FDA-forskrifter er ikke bare avgjørende for produsentenes samsvar, men også for sikkerheten og tilliten til pasienter og brukere.
Vi har diskutert digitalt medisinsk utstyr og relaterte krav flere ganger på QualityMedDev-nettsteder, og dekker ulike emner som f.eks. AI innen medisinsk utstyr, digitale helseprodukterog TGA-tilnærming for regulering av digitalt medisinsk utstyr. Samtidig har FDA publisert ulike retningslinjer i forhold til cybersikkerhet, enten ift krav før markedet og krav etter markedsføring.
FDA har etablert cybersikkerhetskrav som en del av deres regulatoriske tilsyn for å beskytte pasienter og sikre integriteten til medisinsk utstyr. Disse standardene gjelder for hele livssyklusen til en enhet, fra innledende design og utvikling til distribusjon og vedlikehold. Med medisinsk utstyr som blir smartere og mer tilkoblet, blir de stadig mer sårbare for cybertrusler. Integrering av cybersikkerhetspraksis i utviklingsfasen av medisinsk utstyr er et avgjørende skritt for å redusere risikoen ved cyberangrep.
Nøkkelkomponenter i FDAs cybersikkerhetsrammeverk
For å sikre cybersikkerhetsintegriteten til medisinsk utstyr, har FDA skissert krav før markedet som inkluderer behovet for produsenter å innlemme cybersikkerhetstrusselmodell og sikkerhetskontroller fra de tidligste stadiene av enhetens utforming.
Produsenter av utstyr skal etablere og følge kvalitetssystemer for å sikre konsistent samsvar med gjeldende krav og spesifikasjoner for deres produkter. Kravene til disse kvalitetssystemene finnes i Quality System (QS)-forskriften i 21 CFR Part 820 hvis enheten selges i USA, eller andre forskrifter for andre land (f.eks. EU MDR 2017/745 for EU).
Avhengig av enhetens beskaffenhet, kan QS-kravene være relevante under førmarkedsstadiet, postmarkedsstadiet eller begge deler. I forbindelse med forhåndsmarkedsfasen kan det å demonstrere rimelig sikkerhet for sikkerhet og effektivitet for visse enheter med cybersikkerhetsrisiko innebære å inkludere dokumentasjonsutganger knyttet til QS-reguleringen som en del av innsendingen før markedet. For eksempel pålegger ISO 13485:2016 og 21 CFR 820 at produsenter av alle klasser av enheter automatisert med programvare etablerer prosedyrer for å kontrollere utformingen av enheten, for å sikre samsvar med spesifiserte designkrav (referert til som "designkontroller"). Innenfor designkontroller er produsenter pålagt å "etablere og vedlikeholde prosedyrer for å validere enhetsdesignet", som omfatter "programvarevalidering og risikoanalyse, der det er hensiktsmessig". Som en del av den pålagte programvarevalideringen og risikoanalysen kan det hende at produsenter av programvareenheter må innføre risikostyring og valideringsprosesser for nettsikkerhet når det er aktuelt.
Programvarevalidering og risikostyring utgjør avgjørende elementer i cybersikkerhetsanalyser, som avgjør om en enhet gir rimelig sikkerhet for sikkerhet og effektivitet. FDA gir produsenter mandat til å innlemme utviklingsprosesser som vurderer og adresserer programvarerisiko gjennom design- og utviklingsstadiene som en del av designkontrollene. Disse prosessene bør omfatte cybersikkerhetshensyn. Dette inkluderer adressering av identifisering av sikkerhetsrisikoer, etablering av designkrav for å kontrollere disse risikoene, og fremskaffe bevis for at kontrollene fungerer etter hensikten og er effektive i enhetens utpekte miljø, og sikrer tilstrekkelige sikkerhetstiltak.
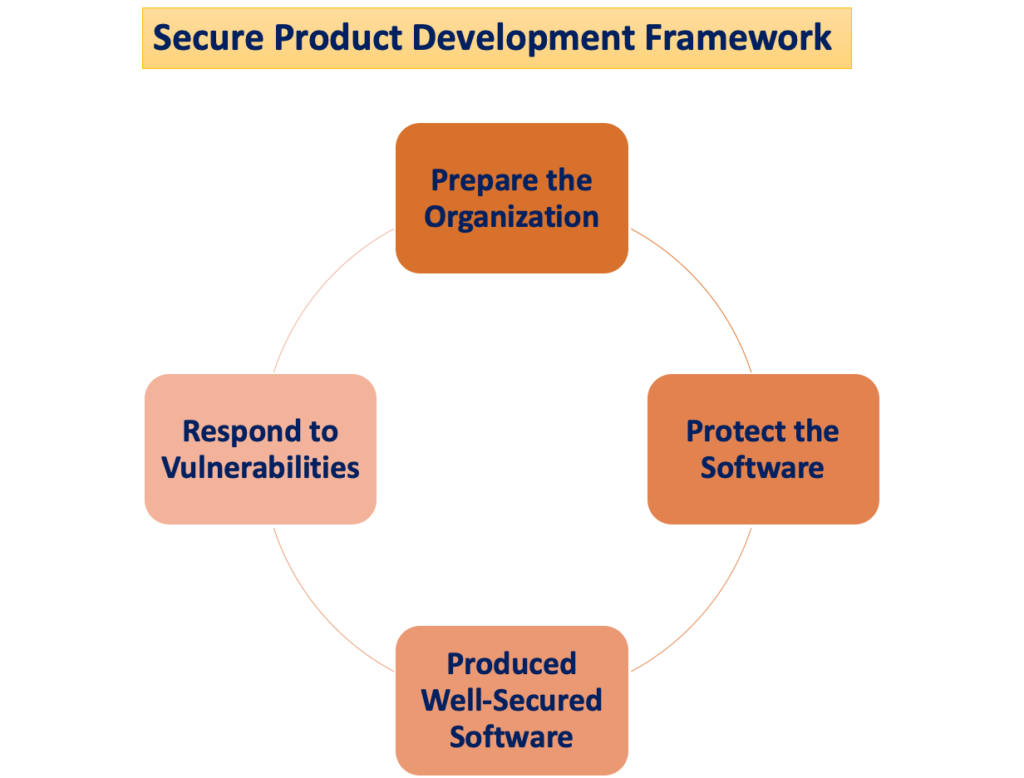
Den "Sikkert rammeverk for produktutvikling"
Potensialet for pasientskade oppstår når cybersikkerhetstrusler utnytter sårbarheter i et system, og hvor lett disse truslene kan kompromittere sikkerheten og effektiviteten til et medisinsk utstyr øker med antallet identifiserte sårbarheter over tid. A Secure Product Development Framework (SPDF) består av prosesser som tar sikte på å identifisere og redusere mengden og alvorlighetsgraden av sårbarheter i produkter. Omfatter alle stadier av et produkts livssyklus – design, utvikling, utgivelse, støtte og dekommisjonering – SPDF er integrert.
Under enhetsdesign kan inkorporering av SPDF-prosesser forhindre nødvendigheten av omstrukturering ved integrering av tilkoblingsbaserte funksjoner etter markedsføring eller adressering av sårbarheter som utgjør ukontrollerte risikoer. Gjennomførbar integrasjon med eksisterende prosesser for produkt- og programvareutvikling, risikostyring og det bredere kvalitetssystemet bidrar til allsidigheten til SPDF.
For å sikre samsvar med kvalitetssystemet (QS) forskriften, anbefales bruk av en SPDF. FDA oppfordrer produsenter til å omfavne SPDF for dens fordeler ved å oppfylle både QS-reguleringen og cybersikkerhetskravene. Det erkjennes imidlertid at alternative tilnærminger også kan tilfredsstille QS-forskriften.
Håndtering av cybersikkerhetsrisikoer
Hovedmålet med å bruke en SPDF (Secure Product Development Framework) er å skape og opprettholde enheter som er både trygge og effektive. Fra et sikkerhetsperspektiv tjener disse enhetene også pålitelighet og motstandskraft. Produsenter og/eller brukere (f.eks. pasienter, helseinstitusjoner) kan deretter administrere disse enhetene, inkludert installasjon, konfigurasjon, oppdateringer og gjennomgang av enhetslogger, gjennom enhetsdesignet og tilhørende merking.
Helsetjenester har muligheten til å administrere disse enhetene innenfor sine egne rammer for styring av cybersikkerhetsrisiko, slik som det anerkjente National Institute of Standards and Technology (NIST) Framework for Improving Critical Infrastructure Cybersecurity, ofte referert til som NIST Cybersecurity Framework eller NIST CSF.
FDA anbefaler at produsenter innlemmer enhetsdesignprosesser, slik som de som er beskrevet i Quality System (QS)-forordningen, for å styrke sikker produktutvikling og vedlikehold. Mens de opprettholder fleksibiliteten for produsenter, kan de også utforske alternative rammer som er i tråd med QS-forordningen og overholder FDA-anbefalingene for implementering av en SPDF. Eksempler inkluderer det medisinske utstyrsspesifikke rammeverket i Medical Device and Health IT Joint Security Plan (JSP) 30 og IEC 81001-5-1. Rammer fra andre sektorer, slik som ANSI/ISA 62443-4-1 Sikkerhet for industriell automasjon og kontrollsystemer Del 4-1: Produktsikkerhetsutvikling livssykluskrav, kan også oppfylle QS-forskrifter.
I de følgende delene av denne artikkelen er det gitt anbefalinger for bruk av SPDF-prosesser, som generelt oppfattes av FDA, som fremhever avgjørende hensyn for å utvikle enheter som er trygge og effektive. Disse prosessene utfyller QS-forordningen, og FDA foreslår at produsenter inkluderer tilsvarende dokumentasjon for gjennomgang i innsendinger før markedet.
Trusselmodell for nettsikkerhet
Trusselmodellering involverer en systematisk prosess for å identifisere sikkerhetsmål, risikoer og sårbarheter i hele det medisinske utstyrssystemet. Deretter innebærer det å definere mottiltak for å forhindre, dempe, overvåke eller svare på virkningene av trusler gjennom hele livssyklusen til det medisinske utstyrssystemet. Når det brukes hensiktsmessig og omfattende, fungerer det som grunnlaget for å optimalisere sikkerheten på tvers av systemkomponenter, produkter, nettverk, applikasjoner og tilkoblinger.
Når det gjelder sikkerhetsrisikostyring, og for å finne passende sikkerhetsrisikoer og kontroller for det medisinske utstyrssystemet, tar FDA til orde for implementering av trusselmodellering for å informere og støtte risikoanalyseaktiviteter. I sammenheng med risikovurdering foreslår FDA å integrere trusselmodellering gjennom hele designprosessen, som omfatter alle elementene i det medisinske utstyrssystemet.
Nøkkelaspekter ved trusselmodellen bør omfatte identifisering av risikoer og reduksjoner, og informere både før og etter reduksjonsrisikoer som vurderes i risikovurderingen for nettsikkerhet. I tillegg bør modellen artikulere antakelser om det medisinske utstyrssystemet eller bruksmiljøet, for eksempel å anta at sykehusnettverk er iboende fiendtlige. Denne forutsetningen ber produsenter om å vurdere scenarier der en motstander kontrollerer nettverket, i stand til å endre, slippe og spille av pakker. Videre bør trusselmodellen fange opp cybersikkerhetsrisikoer introdusert gjennom forsyningskjeden, produksjon, distribusjon, interoperasjon med andre enheter, vedlikeholds-/oppdateringsaktiviteter og dekommisjoneringsaktiviteter, som kan overses i en tradisjonell sikkerhetsrisikovurderingsprosess.
For innsendinger før markedet anbefaler FDA å inkludere trusselmodelleringsdokumentasjon for å vise frem analysen av det medisinske utstyrssystemet, og identifisere potensielle sikkerhetsrisikoer som kan påvirke sikkerhet og effektivitet. Produsenter har fleksibilitet til å velge mellom ulike metoder eller kombinasjoner av metoder for trusselmodellering, og begrunnelsen for deres valgte metodikk bør følge med dokumentasjonen.
Det anbefales å utføre trusselmodelleringsaktiviteter under designgjennomganger, og dokumentasjonen bør gi tilstrekkelig informasjon til at FDA kan vurdere og vurdere sikkerhetsfunksjonene som er integrert i enheten. Denne helhetlige evalueringen bør vurdere både enheten og det bredere systemet den opererer i, med vekt på sikkerheten og effektiviteten til enheten.
Hovedelementene i cybersikkerhetstrusselmodellene kan oppsummeres som følger:
Identifisere potensielle trusler
Utviklingen av en trusselmodell fungerer som et grunnleggende skritt i cybersikkerhetsprosessen, og gjør det mulig for produsenter å identifisere og forstå potensielle cybersikkerhetstrusler som er spesifikke for deres medisinske utstyr. Utviklingen av en trusselmodell fungerer som et grunnleggende skritt i cybersikkerhetsprosessen, og gjør det mulig for produsenter å identifisere og forstå potensielle cybersikkerhetstrusler som er spesifikke for deres medisinske utstyr. Utviklingen av en trusselmodell fungerer som et grunnleggende skritt i cybersikkerhetsprosessen, og gjør det mulig for produsenter å identifisere og forstå potensielle cybersikkerhetstrusler som er spesifikke for deres medisinske utstyr.
Risikovurdering og styring
Risikovurdering og -styring innebærer å evaluere de identifiserte truslene for å fastslå deres potensielle innvirkning og utarbeide passende strategier for å redusere disse risikoene effektivt.
Implementering av sikkerhetskontroller
Sikkerhetskontroller er sikkerhetstiltakene eller mottiltakene som er implementert for å beskytte konfidensialiteten, integriteten og tilgjengeligheten til det medisinske utstyret mot identifiserte trusler.
Fremtidige trender i FDAs retningslinjer for cybersikkerhet
Etter hvert som trusler og teknologi utvikler seg, vil også FDAs retningslinjer for nettsikkerhet utvikle seg. Det er viktig for produsenter å holde seg informerte og smidige i møte med disse endringene. Produsenter bør forutse oppdateringer av regelverk ved å holde seg à jour med bransjetrender og opprettholde en proaktiv tilnærming til cybersikkerhet.
Forberedelse til uforutsette utfordringer er nøkkelen; Etablering av robuste sikkerhetsprotokoller og fremtidsrettede strategier vil posisjonere produsenter til å svare effektivt på fremtidens cybersikkerhetsforskrifter.
Cybersikkerhet er et aspekt ved regulering av medisinsk utstyr som ikke kan overvurderes – det er like iboende for enhetssikkerhet som enhver mekanisk eller elektrisk funksjon. FDA har anerkjent dette, og ved å implementere et omfattende cybersikkerhetsrammeverk, har det som mål å holde tritt med innovasjon og samtidig beskytte folkehelsen. Produsenter, helsepersonell og pasienter må alle samarbeide for å opprettholde disse standardene og sikre fortsatt pålitelighet og sikkerhet til medisinsk utstyr i møte med cybertrusler.
Abonner på QualityMedDev Nyhetsbrev
QualityMedDev er en nettbasert plattform fokusert på kvalitet og regulatoriske emner for medisinsk utstyrsvirksomhet; Følg oss på Linkedin og Twitter for å holde deg oppdatert med de viktigste nyhetene på Reguleringsfeltet.
QualityMedDev er en av de største nettbaserte plattformene som støtter virksomheten innen medisinsk utstyr for emner som overholder forskrifter. Vi sørger for regulatoriske konsulenttjenester over et bredt spekter av emner, fra EU MDR & IVDR til ISO 13485, inkludert risikostyring, biokompatibilitet, brukervennlighet og programvareverifisering og validering og generelt støtte ved utarbeidelse av teknisk dokumentasjon for MDR.
Vår søsterplattform QualityMedDev Academy gir muligheten til å følge opplæringskurs på nett og i eget tempo fokusert på temaer for overholdelse av regelverk for medisinsk utstyr. Disse kursene, utviklet i samarbeid med høyt dyktige fagfolk innen medisinsk utstyrssektoren, lar deg eksponentielt øke kompetansen din over et bredt spekter av kvalitets- og regulatoriske emner for medisinsk utstyrsvirksomhet.

Ikke nøl med å abonnere på vårt nyhetsbrev!
- SEO-drevet innhold og PR-distribusjon. Bli forsterket i dag.
- PlatoData.Network Vertical Generative Ai. Styrk deg selv. Tilgang her.
- PlatoAiStream. Web3 Intelligence. Kunnskap forsterket. Tilgang her.
- PlatoESG. Karbon, CleanTech, Energi, Miljø, Solenergi, Avfallshåndtering. Tilgang her.
- PlatoHelse. Bioteknologisk og klinisk etterretning. Tilgang her.
- kilde: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- : har
- :er
- :ikke
- :hvor
- $OPP
- 2016
- 30
- 350
- 820
- 9
- a
- Om oss
- Academy
- erkjente
- tvers
- Aktiviteter
- I tillegg
- adresse
- adressering
- Legger
- overholde
- administrasjon
- rådes
- talsmenn
- smidig
- sikte
- mål
- justere
- Alle
- tillater
- også
- alternativ
- an
- analyser
- analyse
- og
- forutse
- noen
- aktuelt
- søknader
- anvendt
- Påfør
- tilnærming
- tilnærminger
- hensiktsmessig
- hensiktsmessig
- ER
- Artikkel
- AS
- aspektet
- aspekter
- vurdere
- evaluering
- assosiert
- forutsetningen
- antagelser
- forsikring
- At
- Angrep
- Automatisert
- Automatisering
- tilgjengelighet
- BE
- bli
- bli
- vært
- Fordeler
- styrke
- både
- bred
- bredere
- brakte
- virksomhet
- forretningsdrift
- men
- by
- CAN
- kan ikke
- stand
- fangst
- viss
- kjede
- utfordringer
- Endringer
- Velg
- valgt ut
- klasser
- samarbeide
- samarbeid
- COM
- kombinasjoner
- vanligvis
- Kompletter
- samsvar
- komponenter
- omfattende
- kompromiss
- unnfangelse
- bekymringer
- konfidensialitet
- Konfigurasjon
- tilkoblet
- Tilkoblede enheter
- Tilkoblinger
- Vurder
- betraktninger
- ansett
- konsistent
- består
- utgjør
- konsulent
- moderne
- kontekst
- fortsatte
- kontroll
- kontrollerende
- kontroller
- Tilsvarende
- kunne
- land
- kurs
- dekker
- skape
- kritisk
- Kritisk infrastruktur
- avgjørende
- cyber
- Cyber Attacks
- Cybersecurity
- Dato
- definere
- definisjon
- demonstrere
- distribusjon
- utforming
- design prosess
- utpekt
- bestemme
- utviklet
- utvikle
- Utvikling
- enhet
- Enheter
- forskjellig
- digitalt
- avtagende
- diskutere
- dokumentasjon
- slippe
- medikament
- under
- e
- tidligste
- tjene
- lette
- Effektiv
- effektivt
- effektivitet
- enten
- elementer
- omfavne
- streker
- ansette
- muliggjør
- omfatte
- Omfatter
- altomfattende
- oppmuntrer
- sikre
- sikrer
- Miljø
- avgjørende
- etablere
- etablert
- etablere
- Eter (ETH)
- EU
- europeisk
- Den Europeiske Union
- evaluere
- evaluering
- NOEN GANG
- bevis
- utvikle seg
- eksempler
- eksisterende
- Exploit
- utforske
- eksponentielt
- Face
- fasiliteter
- FDA
- gjennomførbart
- Trekk
- Egenskaper
- felt
- fleksibilitet
- fokuserte
- følge
- etter
- følger
- mat
- Food and Drug Administration
- Food and Drug Administration (FDA)
- Til
- teten
- fremtidsrettet
- funnet
- Fundament
- grunn
- Rammeverk
- rammer
- fra
- fullt
- funksjonalitet
- Dess
- framtid
- general
- generelt
- retningslinjer
- skade
- Ha
- Helse
- helsetjenester
- Høy
- Uthev
- svært
- helhetlig
- Hospital
- Men
- HTML
- HTTPS
- Identifikasjon
- identifisert
- identifisere
- identifisering
- if
- Påvirkning
- Konsekvenser
- gjennomføring
- implementert
- implementere
- viktig
- bedre
- in
- inkludere
- inkluderer
- Inkludert
- innlemme
- innlemme
- Øke
- øker
- stadig
- industriell
- industriell automatisering
- industri
- informere
- informasjon
- informert
- Infrastruktur
- iboende
- innledende
- Innovasjon
- installasjon
- f.eks
- Institute
- integrert
- integrert
- Integrering
- integrering
- integritet
- tiltenkt
- inn
- egenverdi
- introdusert
- involvere
- innebærer
- ISO
- IT
- DET ER
- ledd
- Hold
- nøkkel
- merking
- største
- Livssyklus
- Livssyklus
- MailChimp
- Hoved
- vedlikeholde
- opprettholde
- vedlikehold
- administrer
- ledelse
- mandater
- Produsenter
- produksjon
- max bredde
- Kan..
- MDR
- målinger
- mekanisk
- medisinsk
- medisinsk enhet
- medisinsk utstyr
- Møt
- møte
- metoder
- metoder
- kunne
- Minske
- formildende
- dempe risiko
- modell
- modellering
- modeller
- Overvåke
- mer
- mest
- må
- nasjonal
- Natur
- Navigasjon
- nødvendighet
- Trenger
- nettverk
- nettverk
- nyheter
- nst
- Antall
- Målet
- mål
- of
- on
- ONE
- på nett
- bare
- betjene
- opererer
- Drift
- optimalisere
- Alternativ
- or
- Annen
- vår
- skissert
- utganger
- enn
- oppsyn
- egen
- Fred
- pakker
- del
- pasient
- pasienter
- oppfattet
- perspektiv
- fase
- sentral
- fly
- plattform
- plato
- Platon Data Intelligence
- PlatonData
- spiller
- plugg inn
- poserte
- posisjon
- mulighet
- innlegg
- potensiell
- praksis
- forberedelse
- forebygge
- primære
- Proaktiv
- prosedyrer
- prosess
- Prosesser
- Produkt
- produktutvikling
- Produkter
- fagfolk
- ledetekster
- beskytte
- beskytte
- protokoller
- gi
- forutsatt
- gir
- gi
- offentlig
- folkehelsen
- publisert
- kvalitet
- kvantitet
- område
- begrunnelsen
- rimelig
- gjenkjent
- anbefalinger
- anbefales
- anbefaler
- referert
- Regulering
- forskrifter
- regulatorer
- Overholdelse av regelverk
- i slekt
- forhold
- slipp
- pålitelighet
- forbli
- påkrevd
- Krav
- resiliens
- Svare
- anmeldelse
- Anmeldelser
- Rise
- Risiko
- risikovurdering
- risikostyring
- risikoer
- robust
- Rolle
- trygge
- ivaretakelse
- sikringstiltak
- Sikkerhet
- samme
- scenarier
- seksjoner
- sektor
- sektorer
- sikre
- sikkerhet
- Sikkerhetstiltak
- styring av sikkerhetsrisiko
- sikkerhetsrisiko
- serverer
- flere
- alvorlighetsgrad
- bør
- presentere
- søster
- dyktig
- smartere
- So
- Software
- programvareutvikling
- solgt
- spesifikk
- spesifikasjoner
- spesifisert
- Scene
- stadier
- standarder
- Tilstand
- Stater
- opphold
- blir
- Trinn
- strategier
- innsending
- Innsendinger
- abonnere
- I ettertid
- slik
- tilstrekkelig
- foreslår
- egnet
- levere
- forsyningskjeden
- støtte
- Støtte
- system
- Systemer
- Teknisk
- Teknologi
- enn
- Det
- De
- Fremtiden
- deres
- deretter
- Disse
- de
- denne
- De
- trussel
- trusler
- Gjennom
- hele
- tid
- ganger
- til
- også
- temaer
- tradisjonelle
- Kurs
- Trender
- Stol
- troverdighet
- forstå
- uforutsett
- union
- forent
- Forente Stater
- oppdateringer
- opprett~~POS=TRUNC
- URL
- us
- brukervennlighet
- bruk
- bruke
- Brukere
- utnytte
- validere
- validering
- ulike
- Verifisering
- allsidighet
- Sikkerhetsproblemer
- Sårbar
- we
- nettsteder
- når
- om
- hvilken
- mens
- allment
- vil
- med
- innenfor
- WordPress
- WordPress plugin
- du
- Din
- zephyrnet





