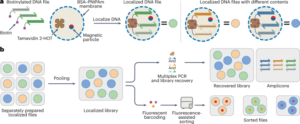
すべての実験と手順は地元の規制委員会と委員会によって承認されており、研究プロトコルに準拠する必要がありました。すべてのマウス手順は、カリフォルニア工科大学動物管理使用委員会 (IACUC; プロトコル 1738) によって承認されたカリフォルニア工科大学で行われました。マーモセット (プロトコル TGC-03) および成ザル (プロトコル LN-14) の手順は NIH で行われ、NIH IACUC によって承認されました。マーモセットの手順はカリフォルニア大学サンディエゴ校 (UCSD) (プロトコル S09147) でも完了しており、UCSD IACUC に準拠し承認されています。乳児マカクの処置は、カリフォルニア大学デービス校のカリフォルニア国立霊長類研究センターで行われ、地元の IACUC (プロトコル 22525) によって承認されました。グリーン モンキーの手順はビルシオで行われ、地元の IACUC によって承認されました。
AAV DNA ライブラリーの生成
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jy89g2w/v1)。以前に詳細に説明したように、最初に DNA レベルで多様性を生成し、次にそれを使用してトランスフェクション材料を作成し、AAV キャプシド ライブラリを作成しました。16。第 588 ラウンドのライブラリーでは、アミノ酸 589 と XNUMX の間に挿入された縮重ヌクレオチドを含むプライマーを使用して、この遺伝的多様性を導入しました。 12,13,16) (VP1 の番号付け; 補足図。 1a)。 7 つの縮重ヌクレオチド ([NNK] × 7) を含むリバースプライマーを使用して、ユニークな 7 mer 配列を含むポリメラーゼ連鎖反応 (PCR) フラグメントをランダムに生成しました。 キャップ 遺伝子。第 66,628 ラウンドの DNA ライブラリーでは、合成オリゴ プール (Twist Bioscience) をリバース プライマーとして使用し、さらなるスクリーニングのために選択されたバリアントのみをコードしました (合計、33,314 個の DNA オリゴ、第 20 ラウンドの選択後に回収された 5 個のバリアントとコドン修飾されたバリアント)それぞれの複製)。すべてのリバースプライマーには、 キャップ これらは、AgeI 制限酵素配列付近の 20 bp 5' オーバーハングを含むフォワード プライマーと対になり、XbaI 制限酵素配列付近に 2621 bp の XNUMX' オーバーハングを含みます。次に、多様な領域を含む PCR フラグメントをギブソン アセンブリ経由で rAAV-ΔCAP-in-cis-Lox プラスミドに挿入し、NEBuilder HiFi DNA アセンブリを使用して、得られた AAV DNA ライブラリ、すなわち rAAV-CAP-in-cis-Lox を生成しました。マスターミックス (New England Biolabs、EXNUMX)。
AAVキャプシドライブラリーの作製
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jyz9g2w/v1)。以前に公開されたプロトコルに従って AAV カプシド ライブラリを生成しました16,70。簡単に説明すると、トランスフェクショングレードの直鎖状ポリエチレンイミン (PEI; Polysciences) を使用して、293 mm 組織培養プレート内の HEK3216T 細胞 (ATCC、カタログ番号 CRL-0063; RRID: CVCL_150) をトランスフェクトしました。各プレートで、1 つのプラスミドをトランスフェクトしました。(2) 組み立てられた rAAV-Cap-in-cis-Lox AAV DNA ライブラリー。AAV のキャプシド化に必要な逆方向末端反復が隣接しています。 (2) AAV9/XNUMX REP-AAP-ΔCAP は、C 末端で AAV 産生に必要な REP および AAP 補足タンパク質をコードします。 キャップ AAV DNA ライブラリーとの組換えとその後の複製能力のある AAV の生成を防ぐために遺伝子が切除されます。 (3) AAV 産生に必要なアデノウイルスタンパク質をコードする pHelper。 (4) pUC18 (Addgene ID: 50004; RRID: Addgene_50004)。哺乳類発現ベクターは含まれませんが、最適な PEI トランスフェクションに適切な窒素対リン酸比を達成するためのフィラー DNA として使用されます。 PEI-DNA 混合物の調製中に、10 mm ディッシュごとに 150 ng の AAV DNA ライブラリー (rAAV-Cap-in-cis-Lox) を追加し、AAV2/9 REP-AAP-ΔCAP、pUC18、および pHelper を混合しました。 1:1:2 の比率 (40 mm ディッシュあたり 150 μg の総 DNA)。トランスフェクションの 60 時間後、ポリエチレングリコール沈殿とイオジキサノール勾配超遠心分離を使用して、細胞ペレットと培地の両方から AAV キャプシド ライブラリを精製しました。次に、定量的 PCR を使用して、確立されたプロトコールに従って直線化されたゲノム標準と比較して DNaseI 耐性ウイルス ゲノムを増幅することにより、AAV キャプシド ライブラリーの力価を決定しました。70.
マーモセットの実験
カプシドライブラリの選択
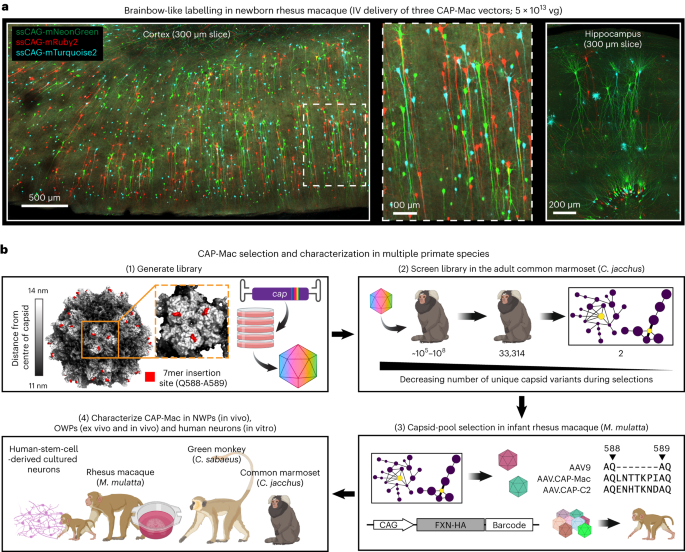
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2)。マーモセット全員 (C.ジャッカス)手順は国立精神衛生研究所(NIMH)で実施され、地元のIACUCによって承認されました。マーモセットはNIMHコロニーで生まれ育ち、27℃、湿度50%の標準条件下で家族グループで飼育されました。彼らは自由に餌を与えられ、NIH の NHP のための霊長類強化プログラムの一環として強化されました。この研究で使用されたすべてのマーモセットについて、IV 注入前の 1:5 血清希釈では検出可能な中和抗体は存在しませんでした (ペンシルベニア大学の Penn Vector Core によって分析)。次いで、それらを数日間個別に収容し、注射前に新しい部屋に順応させた。 2 人の成人男性をライブラリー スクリーニングに使用し、それぞれ 10 人を第 XNUMX ラウンドと第 XNUMX ラウンドのライブラリーに使用しました。注入の前日に、動物の餌を取り除いた。動物を酸素中のイソフルランで麻酔し、大腿静脈上の皮膚を剃り、イソプロパノールスクラブとXNUMX×XNUMX12 AAVキャプシドライブラリーのvgを数分間かけて注入した。麻酔を中止し、動物が活動的になるまで観察し、活動的になるとケージに戻した。次の 3 日間、活動と行動を注意深く監視し、その後は毎日観察しました。
注射の4週間後、マーモセットを安楽死させ(安楽死、VetOne)、1×リン酸緩衝生理食塩水(PBS)で灌流した。第 2 ラウンドのライブラリーの後、脳を 32631 つの冠状ブロックに切断し、80-メチルブタン (Sigma-Aldrich、M80) で急速冷凍し、ドライアイスで冷却し、長期保存のために -XNUMX °C で保存しました。 XNUMX ラウンド目のライブラリーの後、脳を XNUMX つの冠状ブロックに切断し、脊髄および肝臓の切片とともに急速冷凍し、長期保存のために -XNUMX °C で保管しました。
マーモセットにおける AAV の個別の特性評価
このセクションの手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 および https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1)。大人のコモンマーモセット 2 頭 (C.ジャッカス) この実験には、コナン (男性、2.8 歳、0.386 kg) とサンディ (女性、5.8 歳、0.468 kg) が使用されました (補足表) 3 詳細については、を参照してください)。それらを27℃、湿度50%の標準条件下で飼育し、餌と水を自由に摂取させた。すべての動物は集団飼育され、実験は UCSD の皮質システムおよび行動研究所で行われました。すべての実験は UCSD IACUC によって承認されました。注入の前日に、動物の餌を取り除いた。
動物をケタミン(Ketaset、Zoetis 043-304、20 mg kg)で麻酔した。 - 1)、伏在静脈上の皮膚を剃り、イソプロパノールスクラブと2×10で消毒しました。13 vg kg - 1 5分間かけてAAVを注入した。動物は、活動的になるまで監視され、活動的になるとケージに戻されました。次の 5 日間、活動と行動を注意深く監視し、その後は毎日観察しました。血漿中のウイルス濃度を測定するために、3、1、7、14、および21日目に血液サンプルを採取しました。
注射後 31 日目に、前述のようにマーモセットをケタミンで麻酔し、その後安楽死させました (Euthasol、Virbac 200-071、1 ml kg - 1)そして1×PBSで灌流した。脳と臓器を半分に切り、半分を 2-メチルブタン (Sigma-Aldrich、M32631) で急速冷凍し、ドライアイスで冷却し、-80 °C で保存しました。残りの半分を 4% パラホルムアルデヒド (PFA) (Thermo Scientific、J19943-K2) で一晩固定し、PBS アジド (Sigma-Aldrich、S4-2002G、100%) 中で 0.025 °C で保存しました。その後、サンプルは分析のためにカリフォルニア工科大学 (Caltech) に送られました。 GLUT1 染色では、スライスをウサギ抗 GLUT1 (1:200; Millipore-Sigma、カタログ番号 07-1401; RRID: AB_1587074) でインキュベートし、PBS で 1 ~ 200 回洗浄し、ロバ抗ウサギ IgG (711:605) でインキュベートしました。 152; Jackson ImmunoResearch Labs、カタログ番号 2492288-0.1-100; RRID: AB_8787) を使用し、取り付ける前に 10 ~ 017 回洗浄しました。すべての抗体を希釈し、000% Triton X-121 (Sigma-Aldrich、T2337258) および XNUMX% 正常ロバ血清 (Jackson ImmunoResearch Labs、カタログ番号 XNUMX-XNUMX-XNUMX; RRID: AB_XNUMX) を添加した PBS を使用してすべてのインキュベーションを一晩実行しました。室温で振盪しながら。
ウイルスライブラリーの DNA 抽出と NGS サンプルの調製
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2)。我々は以前、TRIzolを使用して水相から核酸を沈殿させることにより、ウイルスライブラリーDNAと内在性宿主RNAを単離できることを報告しました。12,16。したがって、マーモセット組織からウイルスライブラリ DNA を抽出するために、BeadBug (Benchmark Scientific、D100) を使用して、15596 mg の脊髄、肝臓、および脳の各冠状ブロックを TRIzol (Life Technologies、1036) 中でホモジナイズし、マーモセット組織から核酸を単離しました。メーカーの推奨プロトコールに従って水相を調整します。再構成された沈殿をRNase (Invitrogen、AM2288) で処理し、SmaIで消化して、PCRによる下流のウイルスDNAの回収率を向上させました。消化後、メーカーの推奨プロトコールに従って Zymo DNA Clean and Concentrator キット (D4033) で精製し、精製したウイルス DNA を -20 °C で保存しました。
多様化した領域に隣接するイルミナアダプターを付加するために、最初に、抽出されたウイルス DNA の合計の 7% を鋳型として使用して、50 mer 挿入を含む領域を PCR 増幅しました (25 サイクル)。 Zymo DNA 精製後、サンプルを 1:100 に希釈し、7600 サイクルの PCR で可変領域の周囲をさらに増幅し、次の PCR 反応のために結合領域を追加しました。最後に、NEBNext Dual Index Primers (New England Biolabs、E2) を使用して、さらに 16520050 サイクルの PCR によって Illumina フローセルアダプターと独自のインデックスを追加しました。次に、210% 低融点アガロースゲル (Thermo Fisher Scientific、XNUMX) を使用して最終 PCR 産物をゲル精製し、XNUMX bp のバンドを回収しました。
第 2 ラウンドのライブラリーについてのみ、NGS 用にキャプシド化された AAV ライブラリー ssDNA も単離して、ライブラリー濃縮スコアを計算しました。これは、ライブラリー内のさまざまなバリアントの力価の差を正規化するために使用した定量的指標です (参考文献 1 を参照)。 16 および「NGS 読み取りアライメント、分析、およびネットワーク グラフの生成」セクション)。キャプシド化されたウイルスゲノムを単離するために、AAV キャプシド ライブラリを DNaseI で処理し、プロテイナーゼ K を使用してキャプシドを消化しました。次に、フェノール クロロホルムを使用して ssDNA を精製し、2500 つの PCR 増幅ステップでウイルス導入遺伝子を増幅して、Illumina NGS 用のアダプターとインデックスを追加し、精製しました。ゲル電気泳動を使用します。このウイルス ライブラリー DNA は、組織から抽出されたウイルス DNA とともに、Illumina HiSeq XNUMX システム (カリフォルニア工科大学、Millard and Muriel Jacobs Genetics and Genomics Laboratory) を使用したディープ シークエンシングに送られました。
NGS 読み取りアライメント、分析、ネットワーク グラフの生成
NGS 実行からの生の FASTQ ファイルは、カスタム構築されたスクリプト (https://github.com/GradinaruLab/protfarm および https://github.com/GradinaruLab/mCREATE)16。第 33,314 ラウンドのライブラリの場合、これらのデータセットを処理するパイプラインには、低品質のリードを除去するためのフィルタリング、各配列の品質スコアの利用、PCR 誘発変異または高 GC 含有量によるバイアスの除去が含まれていました。フィルタリングされたデータセットは、完全な文字列一致アルゴリズムによって位置合わせされ、位置合わせの品質を向上させるためにトリミングされました。次に、各組織内でのシーケンス実行中の各バリアントの絶対リード数を表示し、脳内で見つかった XNUMX 個のバリアントすべてが第 XNUMX ラウンドの選択に選択されました。
第 2 ラウンドの選択後、同じ分析を実行して、注入されたウイルス ライブラリおよび各組織内の各バリアントのバリアント絶対読み取り数を表示しました。さらに、ライブラリの充実度を計算しました。16 各組織内の各変異について:
$${overline{{rm{RC}}}}_{x,{rm{挿入済み}},{rm{ライブラリ}}}=,frac{{{rm{RC}}}_{x,{rm{注入済み}},{rm{ライブラリ}}}}{mathop{sum }nolimits_{i=1}^{{N}_{{rm{注入済み}},{rm{ライブラリ}}}}{{rm{RC }}}_{i、{rm{挿入済み}、{ライブラリ}}}}、$$
(1)
$${overline{{rm{RC}}}}_{x,{rm{組織}}}=,frac{{{rm{RC}}}_{x,{rm{ウイルス}}}}{mathop {sum }nolimits_{i=1}^{{N}_{{rm{組織}}}}{{rm{RC}}}_{i,{rm{組織}}}},$$
(2)
$${rm{Library},{enrichment}}=,{log }_{10}left(frac{{overline{{rm{RC}}}}_{x,rm{{injected},{library}} }}{{オーバーライン{{rm{RC}}}}_{x,{rm{ティッシュ}}}}右)、$$
(3)
特定のサンプルに対して y (たとえば、注入されたウイルス ライブラリまたは組織サンプル)、RCx,y バリアントの絶対読み取り数です x, Ny は回収されたバリアントの総数であり、 ({overline{{rm{RC}}}}_{x,{y}}) は正規化された読み取りカウントです。
CAP-Mac 配列クラスタリング グラフを構築するために、次の基準に基づいて 1 ラウンド目の NGS データをフィルタリングしました: (100) 注入されたライブラリ サンプルのリード数が 24,186 以上 (33,314/2 バリアント)、(0.7) ライブラリの濃縮度が 415 以上3 つを超える脳サンプル (0.7 バリアント) のスコア、および (0.7) ライブラリー濃縮度が -323 未満の脳サンプル (2 バリアント) よりも、ライブラリー濃縮度が 1 以上の脳サンプルが少なくとも 100 つ多い。 CAP-C2 配列グラフを構築するために、次の基準に基づいて 0.7 ラウンド目の NGS データをフィルタリングしました:(95)注入されたライブラリ サンプルのリード数が XNUMX 以上、(XNUMX)両方のコドン複製が少なくとも XNUMX つの脳サンプルに存在し、 ≥XNUMX のライブラリー濃縮 (XNUMX バリアント)。次に、これらのバリアントを個別に処理して、ペアごとの逆ハミング距離を決定しました (https://github.com/GradinaruLab/mCREATE)、以前に詳しく説明したように、Cytoscape (v. 3.9.0; RRID: SCR_003032) を使用してクラスター化されています。16。提示されたネットワークは、ペアごとの逆ハミング距離が ≥3 の場合、エッジによって接続されたキャプシド バリアント (ノード) を示します。
個々の AAV カプシド バリアントのクローニング
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.n2bvj87ebgk5/v1)。単一変異体の特性評価では、MscI および AgeI を使用して pUCmini-iCAP-PHP.eB (Addgene ID: 103005; RRID: Addgene_103005) バックボーンの修飾バージョンを消化することにより、新しい変異体プラスミドをクローニングしました。各キャプシドバリアントの目的の 100 bp 挿入と、消化されたバックボーンと約 21 bp 重複する AAV9 テンプレートに相補的な領域を含む 20 bp プライマーを設計しました。次に、反応混合物中で 5 μl の 200 nM プライマーと 30 ng の消化されたバックボーンを組み合わせた NEBuilder HiFi DNA Assembly Master Mix を使用してバリアント プラスミドを組み立てました。 AAV.CAP-Mac の生成に使用されるキャプシド プラスミドは、Addgene で入手できます (Addgene ID: 200658; RRID: Addgene_200658)。
個別の AAV の生成と精製
この手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.14egn2dqzg5d/v1)。プールテスト用のバリアントを作成するために、以前に公開されたプロトコルに従いました。70 150 mm 組織培養皿を使用します。 in vivo および in vitro での個々の AAV.CAP-Mac および AAV9 の特性評価では、3320 層 CellSTACK (Corning、150) を利用して高力価でウイルスを効率的に産生し、アカゲザルおよびミドリザルに投与する公開プロトコルを採用しました。具体的には、トランスフェクションの70時間前に、約24%コンフルエントの150 mmディッシュ72枚を150層CellSTACKに継代しました。トランスフェクション当日、120 枚の 15575020 mm ディッシュ用に PEI-DNA トランスフェクション混合物を調製し、トランスフェクション混合物を培地と組み合わせて、CellSTACK の培地を完全に交換しました。 10 mm ディッシュでの生産と同様に、トランスフェクションの 37 時間後に培地を収集して交換しました。トランスフェクションの 20 時間後、エチレンジアミン四酢酸 (Invitrogen、XNUMX) を最終濃度 XNUMX mM になるように加え、CellSTACK の側面を時々旋回させたり叩いたりしながら、XNUMX °C で XNUMX 分間インキュベートして細胞を剥離しました。次に、培地と細胞混合物を除去し、AAV 精製プロトコルに進みました。70。注目すべきことに、超遠心後のバッファー交換ステップでは、Amicon 濾過装置の代わりにポリエーテルスルホン膜を備えた遠心タンパク質濃縮器 (Thermo Scientific、88533) を使用し、0.001% Pluronic F-68 を添加したダルベッコ PBS (Gibco、24040032) を使用しました。
げっ歯類の実験
すべてのげっ歯類の処置はカリフォルニア工科大学で実施され、地元の IACUC によって承認されました。 C57BL/6J (系統番号: 000664; RRID: IMSR_JAX:000664)、BALB/cJ (系統番号: 000651; RRID: IMSR_JAX:000651)、および DBA/2J (系統番号: 000671; RRID: IMSR_JAX:000671) マウスを購入しました。 (すべて雄、生後6〜8週目)ジャクソン研究所産。マウスへの IV 投与の場合、5 × 10 個を投与しました。11 眼窩後洞を通るウイルスの vg70,71 31 ゲージのインスリン注射器 (BD、328438) を使用します。マウスへの AAV の眼窩後注射の詳細については、protocols.io を参照してください (https://doi.org/10.17504/protocols.io.3byl4joy8lo5/v1)。マウスの脳室内(ICV)投与では、5.0×10 を注射しました。10 または1.5×1011 側脳室へのvg。簡単に説明すると、5% O を含むイソフルラン (導入に 1%、維持に 3 ~ 95%) を使用してマウスを麻酔しました。2/ 5%CO2 (1分 - 1)そしてマウスを定位固定フレームに頭部を固定した。頭を剃り、その部分をクロロヘキシジンで消毒した後、0.05 mg ml 2.5 ml を皮下投与しました。 - 1 ブピバカインを投与し、正中線切開を行い、頭蓋骨から血液と結合組織を除去しました。頭部を水平にした後、側脳室の上(ブレグマ後方0.60mm、正中線から1.15mm)に両側にバリ穴を開けた。ウイルスベクターを、0.60ゲージの微量注入針を使用して1.15μl NanoFilシリンジ(World Precision Instruments)に吸引し、針をゆっくりと側脳室(軟膜表面から10mm)まで下げた。針を所定の位置に約 33 分間放置し、マイクロシリンジ ポンプ (World Precision Instruments、UMP1.6) とポンプ コントローラー (World Precision Instruments、Mircro5) を使用して 3 ~ 5 μl のウイルス ベクターを 3 nl 分の速度で注入しました。 - 1。すべてのマウスに術中に 1 mg kg を投与しました - 1 ブプレノルフィン SR および 5 mg kg - 1 ケトプロフェンの皮下投与および30 mg kg - 1 イブプロフェンと60 mg kg - 1 手術後5日間のトリメトプリム/スルファメトキサゾール。マウスへの AAV の ICV 注射の詳細については、protocols.io を参照してください (https://doi.org/10.17504/protocols.io.5qpvorm4dv4o/v1)。発現の 3 週間後、すべてのマウスを PBS で灌流し、4% PFA で固定しました。すべての臓器を抽出し、4.00% PFA 中で一晩インキュベートし、0.01% アジ化ナトリウムを補充した PBS に移し、長期保存のために 4 °C で保存しました。ビブラトーム(Leica Biosystems、VT100S)を使用して脳を1200μmの切片にスライスし、Prolong Diamond Antifade(Invitrogen、P36970)にマウントし、ZEN(Black edition)を使用して共焦点顕微鏡(Zeiss、LSM 880)を使用して画像化しました。組織の取り扱いの詳細については、protocols.io を参照してください (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 および https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1).
アカゲザルの実験
このセクションの手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1)。新生児マカク (0.45 ~ 1.40 kg) は出生時に乳離れしました。最初の 8 か月以内に、マカクザルに AAV ベクターを静脈内またはくも膜下腔内に注入しました。この研究に含まれたすべての成ザル (17 ~ 4.65 歳、11 ~ 0.10 kg) には、IV 投与のみで AAV が注入されました。 IV注射の場合、動物をケタミン(0.10ml)で麻酔し、伏在静脈上の皮膚を剃って消毒した。 AAV(2×10の間)13 および1×1014 vg kg - 1)を0.75ml未満のPBS中で約1分間かけて伏在静脈にゆっくり注入した。 ICM注射の場合、動物に鎮静剤を筋肉内投与し、首の皮膚領域を剃って無菌的に準備した。注射された液体の量に比例して少量のCSFを除去するために、針が大槽内に進められた。次に、AAV の滅菌製剤 (1 × 0.7512 または2.5×1013 vg kg - 1) 収集した液体の量に比例した液体を無菌的に取り付け、ゆっくりと注入しました。すべての動物を、鎮静からの回復中に一日中監視し、その後は有害な所見がないか毎日監視した。すべてのサルは、同種のサルの視界と音が聞こえる範囲内で個別に飼育されました。注射後 4 ~ 11 週間後に組織を収集しました。カリフォルニア国立霊長類研究センターの動物の人道的安楽死に関するガイドラインに従って、動物に深く麻酔をかけ、ペントバルビタールナトリウムを投与した。アカゲザルに注入されたすべての物質にはエンドトキシンが含まれていませんでした(<0.1 EU ml - 1)、タンパク質の純度はドデシル硫酸ナトリウム - ポリアクリルアミドゲル電気泳動によって確認されました。補足表 4 および 5 各実験の投与経路、AAV バリアント、ウイルス量、遺伝子積荷、発現期間をリストします。
アカゲザルのプール検査
このセクションの手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1, https://doi.org/10.17504/protocols.io.3byl4jo68lo5/v1 および https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1)。新生マカクのプール実験 (RM-001 ~ RM-004) は、カリフォルニア大学デービス校の CNPRC で実施され、地元の IACUC によって承認されました。成体マカクのプール実験 (RMN-001 および RMN-002) は NIMH で実施され、地元の IACUC によって承認されました。マカクザルを氷冷したRNaseフリーのPBSで灌流した。灌流時に、脳の一方の半球を急速冷凍し、もう一方の半球を 4 mm の冠状ブロックに切断し、4% PFA で 48 時間後固定し、さらなる処理のために Caltech に移しました。 HA 染色では、スライスをウサギ抗 HA (1:200; Cell Signaling Technology、カタログ番号 3724; RRID: AB_1549585) でインキュベートし、PBS で 1 ~ 200 回洗浄し、ロバ抗ウサギ IgG (711:605; 152:2492288; RRID: AB_0.1) でインキュベートしました。 Jackson ImmunoResearch Labs、カタログ番号 100-8787-10; RRID: AB_017)、実装前に 000 ~ 121 回洗浄しました。すべての抗体を希釈し、2337258% Triton X-XNUMX (Sigma-Aldrich、TXNUMX) および XNUMX% 正常ロバ血清 (Jackson ImmunoResearch Labs、カタログ番号 XNUMX-XNUMX-XNUMX; RRID: AB_XNUMX) を添加した PBS を使用してすべてのインキュベーションを実行しました。室温で振盪しながら一晩。
ウイルス DNA と全 RNA を単離するために、脳と肝臓からの 100 mg のスライスを、BeadBug (Benchmark Scientific、D15596) を使用して TRIzol (Life Technologies、1036) 中でホモジナイズし、メーカーの推奨プロトコールに従って全 DNA と RNA を回収しました。 。回収した DNA を RNase で処理し、SmaI で制限消化し、Zymo DNA Clean and Concentrator キット (D4033) で精製しました。回収した RNA を DNase で処理し、製造元の推奨プロトコールに従って SuperScript III (Thermo Fisher Scientific、18080093) およびオリゴ (dT) プライマーを使用して mRNA から cDNA を生成しました。 50 ng のウイルス DNA または cDNA をテンプレートとして使用し、PCR を使用してバーコード領域を増幅しました。 Zymo DNA 精製後、サンプルを 1:100 に希釈し、Illumina NGS 用のアダプターを付加するプライマーを使用してバーコード領域をさらに増幅しました。クリーンアップ後、これらの産物を、Illumina シーケンシング用 NEBNext Dual Index Primers (New England Biolabs、E7600) を 2 サイクル使用してさらに増幅しました。次に、16520050% 低融点アガロースゲル (Thermo Fisher Scientific、XNUMX) を使用して最終 PCR 産物をゲル精製しました。プール テストの強化はライブラリの強化と同じように計算されましたが、図に示されています。 2b、c 線形スケールで。
アカゲザルにおける CAP-Mac の個別の特徴付け
このセクションの手順の詳細は、protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 および https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1)。新生マカクザルに PBS と 4% PFA を灌流しました。脳を4mmの冠状ブロックに切片化し、すべての組織を4%PFA中で3日間後固定し、その後PBS中で保存した。 in vivo での個体特性評価に使用した 4 匹の成ザル (RM-4、3 歳、020 kg) を RNase フリーの PBS で灌流し、半球の 17 つを急速冷凍し、もう 11 つを 4 mm の冠状ブロックに分割しました。 4% PFA で後固定。すべての組織はさらなる処理のためにカリフォルニア工科大学に移送されました。ビブラトームを使用して脳と肝臓を 100 μm のスライスに切片化しました。さらに、脳および脊髄の切片を30%スクロース中で一晩インキュベートし、OCT化合物(Scigen、4586)に包埋し、クライオスタット(Leica Biosystems、CM50)を使用して1950μmのスライスに切片化した。すべてのスライスは Prolong Diamond Antifade を使用してマウントされ、共焦点顕微鏡を使用して画像化されました。髄腔内投与されたマカクザルの脊髄および脳切片の GFP 染色では、切片をニワトリ抗 GFP (1:500; Aves Labs、カタログ番号 GFP-1020; RRID: AB_10000240) とインキュベートし、PBS で 1 ~ 200 回洗浄しました。 、ロバ抗ニワトリ IgY (703:605; Jackson ImmunoResearch Lab、カタログ番号 155-2340379-0.1; RRID: AB_100) とインキュベートし、マウントする前に 8787 ~ 10 回洗浄しました。すべての抗体を希釈し、017% Triton X-000 (Sigma-Aldrich、T121) および XNUMX% 正常ロバ血清 (Jackson ImmunoResearch、XNUMX-XNUMX-XNUMX) を添加した PBS を使用してすべてのインキュベーションを室温で振盪しながら一晩実行しました。
形態学的再構築のために、脳を 300 μm の切片に分割し、屈折率が一致する溶液中でインキュベートしました。72 屈折率整合溶液に浸漬したスライドにマウントする前に、72 時間放置します。共焦点顕微鏡と 25 倍の対物レンズ (LD LCI Plan-Apochromat ×25/0.8 Imm Corr DIC) を使用し、液浸液として 100% グリセロールを使用して画像化しました。タイルをキャプチャしました Z 対象のセルの周囲にスタック (推奨されるキャプチャ設定を使用して各フレームに 1,024 × 1,024) を配置し、トレース用に適切な視野をトリミングします。トレースは、Imaris (Oxford Instruments; RRID: SCR_007370) で半自動および自動の方法を使用して実行されました。
ニューロン (NeuN) および星状細胞 (S100β) の定量化のために、抗 NeuN (EPR12763) 抗体 (1:200; Abcam、カタログ番号 ab177487; RRID: AB_2532109) または抗 S100β 抗体 (1:200; Abcam) を使用してスライスを染色しました。 、カタログ番号ab52642;RRID:AB_882426)0.1%トリトンX-100および10%正常ロバ血清を補充したPBS中で一晩。スライスを PBS で 52642 ~ 882426 回洗浄し、PBS + 0.1% 中の Alexa Fluor 100 (10:647; Jackson ImmunoResearch Labs、カタログ番号 1-200-711; RRID: AB_605) と結合した抗ウサギ IgG 抗体中で一晩インキュベートしました。 Triton X-152 + 2492288% 通常のロバ血清。 0.1〜100回洗浄し、Prolong Diamond Antifadeを使用して取り付けた後、次の結果が得られました。 Z 共焦点顕微鏡と25倍の対物レンズを使用してスタックします。 Python のカスタム スクリプト (RRID: SCR_008394) と Cellpose (https://www.cellpose.org/; RRID: SCR_021716)73.
体外二光子イメージング
公開されたプロトコルで以前に説明されているように、イメージングに適したサイズの脳スライスをビブラトームを使用して大きなスライスから 400 μm の厚さで調製し、二光子イメージングの前にカルボゲンガスで泡立てた人工脳脊髄液に保存しました。74,75。 GCaMP8 応答をテストするために、画像化されたニューロンから 4 ~ 5 μm 離れた場所に配置された細胞外単極電極を使用して、指定されたパルス数の電気刺激 (80 ~ 0.3 V、100 Hz、持続時間 200 秒) が送達されました。撮像のフレームレートは30Hzとした。セグメント化された関心領域のトレースは Δ としてプロットされました。F/F0 = (F(t)− F0)/F0ここで、 F0 は、電気刺激前のすべての蛍光値の平均として定義されます。立ち上がり時間は、信号の立ち上がり位相がピークの 10% からピークの 90% に達するまでに必要な時間として定義されました。減衰時定数は、指数関数の合計を信号の減衰位相に当てはめることによって得られます。シグナル対ノイズ比は、電気刺激前のシグナルのピーク振幅を蛍光トレースの標準偏差で割ることによって得られました。
成体アカゲザルスライスの特性評価
ワシントン国立霊長類研究センターからのアカゲザル成体 14 頭 (1 歳 10.83 か月、体重 300 kg) は定期的な安楽死が計画されており、脳は施設の組織配布プログラムの一環として収集されました。上側頭回のブロックを XNUMX μm のスライスに切断し、スライスを回収しました。74 前述したように、気液膜界面で培養されます。76。スライスを播種してから約 30 分後、1 ~ 2 μl の AAV (5 × 1013 vg ml - 1 ssCAG-FXN-HA または ssCAG-eGFP のいずれかをパッケージ化した AAV9 または AAV.CAP-Mac)。実験は各条件について生物学的三重反復で実施し、形質導入後48日目の組織収集まで8時間ごとに培地を交換した。組織収集の日に、形質導入を確認するためにスライスを画像化し、スライスを半分に切り、各半分のスライスをドライアイス-エタノール浴で急速冷凍しました。サンプルはさらに処理するまで -20 °C で保管されました。
各半分のスライスを処理しました (DNA および RNA の回収用に 69504 つずつ)。 Qiagen DNeasy Blood and Tissue キット (Qiagen、カタログ番号 15596026) を使用して DNA を単離し、TRIzol (Thermo Fisher Scientific、カタログ番号 12183018) および PureLink RNA Mini キット (Thermo Fisher Scientific、カタログ番号 350A) を使用して RNA を回収しました。 PureLink RNA Mini キットの最初の洗浄を次のように変更することにより、RNA サンプルから DNA を除去しました: 1 μl の洗浄バッファー 80 で洗浄し、次に RDD バッファー (Qiagen カタログ番号 79254) 中の 15 μl の RNase フリー DNaseI を加え、インキュベートします。カラムを室温で 350 分間放置します。次に、プロトコールを続行する前に、1 μl の洗浄バッファー 400 で再度洗浄します。 Promega GoScript Reverse Transcription キット (Promega、カタログ番号 A20) を使用して、5000 μl 反応で XNUMX ng の全 RNA から第一鎖 cDNA 合成を実行しました。
次に、Roche Lightcycler II での定量的 PCR を使用して、各サンプルで見つかったベクター ゲノムとウイルス転写物を評価しました。ここでは、Thermo Fisher Scientific の TaqMan プローブ (EGFP-FAM プローブ、アッセイ ID Mr100_mr、カタログ番号 20; カスタム ゲノム参照プローブ CN04097229-4331182-VIC、アッセイ ID ARH2386DUK、カタログ番号) を使用した 2 µl の増幅反応で 6 ng の DNA を使用しました。 4448512、両方をターゲットにするように設計されています M. ムラッタ および マカカネメストリーナ).
ミドリザルの実験
オール・ザ・グリーン・モンキー (C.サバエウス) 手順は Virscio で実施され、IACUC によって承認されました。すべてのサルは中和抗体についてスクリーニングされ、力価が 1:5 未満であることが確認されました。生後約 7 ~ 8 か月(1.0 ~ 1.3 kg)のサルに静脈内投与が行われました(補足表) 6)。用量配合物は、投与前少なくとも10分間、ただし60分以内にほぼ室温まで平衡化させた。 IV用量は、0日目の体重に基づいた。動物をケタミン(10 mg kg)で鎮静させた。 - 1)およびキシラジン(1.6 mg kg - 1)。注射領域を剃り、クロロヘキシジンおよび70%イソプロパノールで下処理し、IVカテーテルの挿入前に外科的にこすった。投与は AAV の 7.5 回の IV 注入で行われました (10 × XNUMX13 または7.6×1013 vg kg - 1) 0 日目に、手持ち式注入装置を使用して伏在静脈経由で目標速度 1 ml/分で投与 - 1。全体的な健康状態は、投与の1週間前からケージ側で観察することにより1日2回確認した。予定された安楽死時間に、サルはケタミン(1~8 mg kg)で鎮静されました。 - 1 筋肉内)およびペントバルビタールナトリウム(100 mg kg)で安楽死させた - 1 IVから効果)。角膜反射が消失した場合、毎分約 100 ml の速度に設定された蠕動ポンプを使用して、冷却 PBS を使用して経心臓灌流 (左心室) を実行しました。 - 1 組織採取前に、漏れ出た液体が透明になるまで続けてください。組織の立方体が左脳半球および他のさまざまな臓器から収集され、生体内分布のためのさらなる処理のために液体窒素の気相中で凍結されました。右脳半球を取り出し、約 4 mm の冠状スライスに切断し、約 20 倍量の 10% 中性緩衝ホルマリンで無傷のまま室温で約 24 時間後固定しました。
Thermo Fisher MagMax DNA Ultra 2.0 抽出キット (カタログ番号 A36570) を使用して、CNS および末梢組織からゲノム DNA を抽出しました。 DNA の収量は、Qubit dsDNA アッセイによる蛍光定量によって評価されました。約 20 ng の DNA を各 20 μl 反応液にロードし、プレートを BioRad CFX Connect リアルタイム PCR 検出システム (カタログ番号 1855201) で実行しました。ウイルスコピー数アッセイの特異性は、単一の増幅産物の検出によって検証されました。検出下限を評価して反応あたり XNUMX コピーを超える感度。検量線を確保することで直線性を向上 R2 >0.95でした。反応は FastStart Universal SYBR Green Master (Rox) (カタログ カタログ番号 4913850001) で組み立てました。プライマーの配列は、ACGACTTCTTCAAGTCCGCC (順方向) および TCTTGTAGTTGCCGTCGTCC (逆方向) でした。 PCR プロトコールでは、95 °C で 180 秒の初期変性ステップを使用し、続いて 40 °C で 95 秒および 15 °C で 60 秒の 60 サイクルを使用し、各 60 °C サイクルの後にイメージングステップを実行しました。標準曲線は、ウイルス内に存在する 1 × 10 個の GFP テンプレート配列を含む直線化プラスミドを使用して作成されました。8 ~1×1010 マトリックス効果を制御するために、この研究のサンプルと同じキットを使用して調製されたナイーブ未処理マカク DNA サンプルで希釈したコピー。ウイルス DNA のコピーは、最も適合する直線の方程式を使用して標準曲線から計算されました。感染多重度の値は、反応ごとに測定された宿主細胞 DNA の総ゲノム重量に基づいて計算されました。
固定後、組織を 10% > 20% > 30% スクロースに 24 °C で 4 時間ずつ置き、次に OCT コンパウンドに包埋し、凍結切片を作成するまで -80 °C で保存しました。組織ブロックは、20 μm スライスに切片化する前にクライオスタット内で -30 °C に加熱され、凍結切片化後にスライド上にドライマウントされました。切片化後、スライドを室温で一晩放置して乾燥させた。ニューロンの定量化を支援するために、以下の抗体および濃度で切片を染色しました:ウサギ抗 GFP (1:100; Millipore-Sigma、カタログ番号 AB3080; RRID: AB_91337) およびマウス抗 NeuN (A60) (1:500; Millipore-Sigma、カタログ番号 MAB377; RRID: AB_2298772)。二次抗体染色には、次の二次抗体と濃度を使用しました: ロバ抗ウサギ Alexa Fluor 488 (1:500; Thermo Fisher Scientific、カタログ番号 A-21206; RRID: AB_2535792) およびロバ抗マウス Alexa Fluor 647 (1:500) :31571; Thermo Fisher Scientific、カタログ番号 A-162542; RRID: AB_1)。全ての抗体を、0.25%トリトンX-100(PBST)および5.00%正常ロバ血清を補充した1×PBSで希釈した。一次抗体のインキュベーションを室温で一晩放置した。次いで、切片をPBSTで洗浄した。二次抗体のインキュベーションを室温で 0.25 時間実施しました。切片をPBSTで3回洗浄した。切片をDAPI溶液(100:5.00; Invitrogen、D2)中で室温で1分間インキュベートし、その後洗浄した。 Prolong Diamond Antifadeを使用して切片にカバースリップをかけました。
動物あたり 800 つの切片を染色し、画像化しました。各セクションは 1 回撮影され、各関心領域には合計 500 枚の画像が含まれます。関心のある組織領域は、次の取得パラメータを使用して Keyence BZ-X5 で画像化されました: GFP (1/1 秒)、Cy12 (XNUMX 秒)、DAPI (XNUMX/XNUMX 秒)、高解像度 Z 1.2μmピッチで積層します。以下の脳の小領域を画像化した:前頭葉皮質、頭頂葉皮質、側頭葉皮質、後頭皮質、小脳、尾状核、被殻および視床(内側核、腹側外側核、および腹側後核)。定量化のために ImageJ (RRID: SCR_003070) を使用して、半自動細胞計数法を実行しました。閾値と粒子分析を使用して、NeuN 陽性細胞と DAPI 陽性細胞を定量しました。 ImageJ のセルカウンターを使用して、GFP 陽性細胞、および GFP と NeuN の二重陽性細胞を手動でカウントしました。
iPS細胞実験
神経培養物は、メーカーのプロトコルに従って、Stemdiff Forebrain Differentiation and Maturation キット (それぞれ StemCell # 08600 および # 08605) を使用して iPSC 由来の神経前駆細胞を分化および成熟させることによって生成されました。神経前駆細胞は、包皮線維芽細胞由来 iPSC 株:ACS-1019 (ATCC# DYS-0100; RRID: CVCL_X499)、Stemdiff SMADi Neural Induction kits (StemCell l#08581)、Stemdiff Neural Rosette Selection による選択の分化によって生成されました。メーカーのプロトコルに従って、試薬 (StemCell I#05832) および Stemdiff Neural Progenitor Media (StemCell I#05833) での増殖。ニューロンは、形質導入のために再播種する前に少なくとも 8 日間成熟させました。
ポリオルニチンおよびラミニンでコーティングされた黒色壁の15,000ウェル光学プレートにウェルあたり96細胞を播種した成熟ニューロン培養物を、形質導入前にさらに4日間培養した。複製ウェルに、90% 成熟培地および 10% OptiPRO SFM で 4 桁にわたって連続希釈したウイルスを形質導入しました。形質導入の 1 日後、培養物を XNUMX% PFA で固定し、XNUMX μg ml で対比染色しました。 - 1 Hoechst 33322。形質導入細胞の同定は、CellInsight CX60 HCS プラットフォーム上の 386 チャンネル蛍光検出 (Hoechst、ex440/em485; eGFP、ex521/em5) を使用して、ウェルあたり XNUMX フィールドをイメージングすることによって決定されました。個々の細胞は、核のヘキスト検出と、サイズと接触に制約されたリング マスクを各細胞に適用することによって識別されました。細胞形質導入は、個々のリングマスク内の閾値レベルを超える eGFP 蛍光を測定することによって決定されました。各集団について、形質導入された細胞の割合を適用用量に対してプロットしました。カーブフィットとEC50 値は、Prism GraphPad (RRID: SCR_002798) アゴニスト対応答 (5,000 パラメーター) 回帰法を使用して決定されました。細胞あたりの eGFP 発現効率を報告するために、ウェルあたり最低 XNUMX 個の細胞にわたって各リング マスクから eGFP スポット蛍光強度を平均しました。曲線の当てはめは、濃度回帰法として Prism GraphPad Biphasic X を使用して得られました。
統計と再現性
代表的な画像の場合、各サンプルから少なくとも 2.38 つの別々のスライスがイメージング用にマウントされました。 2.38 頭の動物の各脳領域内で、少なくとも 50 つの異なる視野が撮影されました (タイリング後の最小視野、XNUMX mm × XNUMX mm、スライス厚、XNUMX μm)。これは、XNUMX つの脳スライスにわたる XNUMX つの別々の視野に相当します。 、画像サンプル間で一貫性を確保するため。
レポートの概要
研究デザインの詳細については、 ネイチャー ポートフォリオ レポートの概要 この記事にリンクされています。
- SEO を活用したコンテンツと PR 配信。 今日増幅されます。
- PlatoData.Network 垂直生成 Ai。 自分自身に力を与えましょう。 こちらからアクセスしてください。
- プラトアイストリーム。 Web3 インテリジェンス。 知識増幅。 こちらからアクセスしてください。
- プラトンESG。 自動車/EV、 カーボン、 クリーンテック、 エネルギー、 環境、 太陽、 廃棄物管理。 こちらからアクセスしてください。
- ブロックオフセット。 環境オフセット所有権の近代化。 こちらからアクセスしてください。
- 情報源: https://www.nature.com/articles/s41565-023-01419-x
- :は
- :どこ
- $UP
- 000
- 1
- 10
- 100
- 11
- 12
- 13
- 14
- 視聴者の38%が
- 16
- 17
- 180
- 2%
- 20
- 200
- 2011
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 24
- 25
- 27
- 30
- 300
- 31
- 33
- 39
- 40
- 50
- 500
- 60
- 66
- 7
- 70
- 72
- 75
- 8
- 80
- 9
- 視聴者の38%が
- a
- 上記の.
- 絶対の
- アクセス
- 従う
- 従った
- 達成する
- 買収
- 越えて
- アクティブ
- アクティビティ
- Ad
- 加えます
- 追加されました
- NEW
- さらに
- 投与
- 管理
- 採択
- 成人
- 高度な
- 不利な
- 後
- 再び
- 年齢
- AL
- アレクサ
- アルゴリズム
- 整列した
- すべて
- 許可されて
- 沿って
- また
- 量
- 増幅
- 増幅した
- 増幅する
- an
- 分析
- アンカー
- および
- 動物
- 動物
- 抗体
- どれか
- 適用された
- 適用
- 適切な
- 承認された
- 約
- AREA
- 周りに
- 記事
- 人工の
- AS
- 組み立て
- アセンブリ
- 評価された
- 評価中
- アシスト
- At
- 自動化
- 利用できます
- 平均
- 離れて
- バックボーン
- BAND
- ベース
- BD
- BE
- になりました
- 開始
- さ
- ベンチマーク
- BEST
- の間に
- バイアス
- 拘束
- ブラック
- ブロック
- ブロック
- 血
- ボディ
- 生まれる
- 両言語で
- BP
- 脳
- 脳
- 簡潔に
- た
- バッファ
- 焙煎が極度に未発達や過発達のコーヒーにて、クロロゲン酸の味わいへの影響は強くなり、金属を思わせる味わいと乾いたマウスフィールを感じさせます。
- by
- ケージ
- 計算する
- 計算された
- カリフォルニア州
- 缶
- キャプチャー
- 捕捉した
- これ
- 貨物
- CAT
- セル
- 細胞
- 細胞の
- センター
- 中央の
- チェーン
- チャンネル
- 変化する
- 変更
- 選ばれた
- クリア
- クリック
- 密接に
- クラスタリング
- コレクション
- コラム
- 組み合わせた
- 結合
- 委員会
- コマンドと
- 補完的
- コンプリート
- 記入済みの
- コンプライアンス
- 従う
- 濃度
- 条件
- 条件
- 確認します
- 確認済み
- お問合せ
- 交流
- 定数
- 構築する
- 含まれている
- 含まれています
- コントロール
- コントローラ
- コピー
- 基本
- コロナル
- カウンター
- 基準
- 文化
- 曲線
- カスタム
- 特注の
- カット
- サイクル
- サイクル
- daily
- データ
- データセット
- デイビス
- 中
- 日
- 深いです
- 定義済みの
- 配信
- 配達
- 記載された
- 設計
- 設計
- 希望
- 細部
- 検出
- 決定する
- 決定
- 偏差
- デバイス
- Devices
- ダイヤモンド
- ディエゴ
- の違い
- 異なります
- 希釈
- ディスプレイ
- 表示される
- 距離
- 明確な
- ディストリビューション
- 多様化した
- 多様性
- DNA
- 行われ
- 線量
- 投与
- ドライブ
- デュレーション
- 間に
- e
- E&T
- 各
- 前
- エディション
- 効果
- 効果
- 効率
- 効率的な
- 効率良く
- どちら
- 要素は
- 排除
- 埋め込まれた
- イングランド
- 確保
- 確保する
- 設立
- エーテル(ETH)
- EU
- 評価
- あらゆる
- 例
- 交換
- 拡大
- 実験
- 実験
- 表現
- エキス
- 抽出
- 家族
- FRBは
- 女性
- フィールド
- フィールズ
- イチジク
- フィギュア
- フィルタリング
- ファイナル
- 最後に
- 調査結果
- 名
- フィット
- フィッティング
- 五
- 固定の
- フラッシュ
- フロー
- 流体
- 続いて
- フォロー中
- 次
- フード
- 収量のために
- フォワード
- 発見
- 4
- FRAME
- 無料版
- から
- 凍結
- 機能的な
- さらに
- GAS
- ゲージ
- 生成する
- 生成された
- 世代
- 遺伝学
- ゲノム
- ゲノミクス
- 与えられた
- グラフ
- 大きい
- グリーン
- グループ
- グループの
- ガイドライン
- 半分
- ハンドリング
- 持ってる
- 持って
- 健康
- こちら
- ハイ
- 高解像度の
- 穴
- 均質化した
- host
- HTTPS
- 人間
- ICE
- ID
- 同一の
- 識別
- 特定され
- if
- ii
- 3
- 画像
- イメージング
- 浸した
- イマージョン
- 改善します
- in
- 含まれました
- 培養
- 単独で
- index
- 示された
- 索引
- 個人
- 個別に
- 誘導
- 感染
- 情報
- 注入された
- 輸液
- 初期
- 当初
- を取得する必要がある者
- 機関
- 制度の
- 楽器
- 関心
- インタフェース
- に
- 静脈内
- 導入
- 関係する
- 分離された
- Jackson (ジェームズ・ジャクソン)
- キット
- ラボ
- 実験室
- ラボ
- より大きい
- ld
- 最低
- 左
- less
- レベル
- ライブラリ
- 図書館
- 生活
- LIMIT
- LINE
- LINK
- リンク
- 液体
- リスト
- 肝臓
- ローカル
- 長期的
- 損失
- 下側
- 下げ
- 製
- メンテナンス
- 手動で
- マッピング
- mask
- マスク
- マスター
- 一致
- マッチング
- 材料
- マトリックス
- だけど
- 測定された
- 計測
- メディア
- ミディアム
- メンタル
- メンタルヘルス
- 方法
- メソッド
- メトリック
- マウス
- 顕微鏡
- 分
- 最小
- 分
- ミックス
- 混合
- ML
- 修正されました
- 監視対象
- 月
- ヶ月
- 他には?
- mRNA
- MSCI
- すなわち
- ナノテクノロジー
- 国民
- 自然
- 近く
- 必要
- ネットワーク
- ネットワーク
- ニューラル
- ニューラル
- ニューロン
- 新作
- 次の
- NIHの
- いいえ
- ノード
- 通常の
- 数
- 客観
- 得
- 発生した
- of
- 古い
- on
- ONE
- の
- 最適な
- 最適化
- or
- 受注
- その他
- 私たちの
- が
- 一晩
- オックスフォード
- 酸素
- 包装
- 対になった
- パラメータ
- 部
- 粒子
- PBS
- PCR法
- ピーク
- ペン
- ペンシルベニア州
- 以下のために
- 割合
- 完璧
- 実行
- 周辺
- 相
- パイプライン
- ピッチ
- 場所
- 計画されました
- プラズマ
- プラットフォーム
- プラトン
- プラトンデータインテリジェンス
- プラトデータ
- さらに
- プール
- 人口
- ポートフォリオ
- 精度
- 準備
- 準備
- 現在
- PLM platform.
- 防ぐ
- 前に
- 主要な
- プライマー
- プローブ
- 手続き
- 手続き
- プロセス
- 処理されました
- 処理
- 作り出す
- 生産された
- プロダクト
- 生産
- 製品
- 前駆細胞
- 演奏曲目
- 保護
- タンパク質
- タンパク質
- プロトコル
- は、大阪で
- 公表
- ポンプ
- 購入した
- Python
- 品質
- キュービット
- ウサギ
- 隆起した
- レート
- 比
- リーチ
- 反応
- 反応
- 読む
- への
- 受け
- 推奨される
- 回復
- 地域
- 地域
- 回帰
- レギュレータ
- 相対
- 削除します
- 削除済み
- 複製
- レポート
- 報告
- 各種レポート作成
- 代表者
- で表さ
- の提出が必要です
- 研究
- それぞれ
- 応答
- 回答
- 結果として
- 逆
- 右
- リング
- 上昇
- 上昇
- RNAを
- 堅牢な
- ロッシュ
- ルーム
- ルート
- ラン
- runs
- s
- 同じ
- サン
- サンディエゴ
- 砂の
- 規模
- 予定の
- SCI
- 科学的な
- スコア
- スコア
- スクリーニング
- スクリプト
- 二次
- セクション
- セクション
- セグメンテーション
- 選択
- 選択
- 選択的
- 感度
- 送信
- 別
- シーケンス
- シーケンシング
- ヒアルロン酸抗酸化セラム
- セッションに
- 設定
- いくつかの
- 出荷
- 表示する
- 側面
- 視力
- シグナル
- 同様の
- 座る
- SIX
- サイズ
- 肌
- スライス
- スライド
- スライド
- ゆっくり
- 小さい
- ナトリウム
- 溶液
- 音
- 特に
- 特異性
- Spot
- スタック
- スタック
- 標準
- 手順
- ステップ
- ストレージ利用料
- 保存され
- 文字列
- 勉強
- それに続きます
- 適当
- 優れた
- 表面
- 合成
- 全身の
- システム
- テーブル
- 撮影
- タップ
- ターゲット
- 対象となります
- ターゲット
- テクノロジー
- テクノロジー
- template
- 10
- ターミナル
- テスト
- より
- それ
- エリア
- アプリ環境に合わせて
- それら
- その後
- そこ。
- したがって、
- ボーマン
- 彼ら
- この
- 三
- しきい値
- 介して
- 全体
- 時間
- <font style="vertical-align: inherit;">回数</font>
- 組織
- 〜へ
- 取った
- トータル
- トレース
- トレーシング
- 転送
- 転送
- 治療
- トリトン
- Twice
- ツイスト
- 2
- 超
- 下
- ユニーク
- ユニバーサル
- 大学
- カリフォルニア大学
- ペンシルベニア大学
- まで
- つかいます
- 中古
- 活用する
- 活用
- 検証済み
- 価値観
- バリアント
- さまざまな
- バージョン
- 対
- 、
- 詳しく見る
- ウイルスの
- ウイルス
- ウイルス
- 生体
- ボリューム
- ました
- ワシントン
- 水
- we
- 週間
- ウィークス
- 重量
- WELL
- ウェルズ
- した
- which
- 全体
- 広範囲
- 以内
- 世界
- X
- 年
- 産出
- 収量
- 禅
- ゼファーネット