Il nuovo articolo affronta gli aspetti relativi alle azioni di esecuzione che l'autorità può intraprendere e descrive anche i vari tipi di norme.

Sommario
La Food and Drug Administration (FDA o Agency), l'autorità di regolamentazione statunitense in materia di prodotti sanitari, ha pubblicato un documento di orientamento dedicato ai dispositivi medicali di imaging a raggi X in termini di conformità agli standard IEC a cui sono soggetti in quanto prodotti elettronici. Il documento descrive l'approccio che l'autorità applicherà nel valutare l'uso degli standard pertinenti, fornisce ulteriori chiarimenti in merito ai requisiti normativi applicabili, nonché raccomandazioni che devono essere seguite dai produttori di dispositivi medici e dalle altre parti coinvolte al fine di garantire la loro conformità. Allo stesso tempo, le disposizioni della guida non sono vincolanti nella loro natura giuridica, né intendono introdurre nuove regole o imporre nuovi obblighi. Inoltre, potrebbe essere applicato un approccio alternativo, a condizione che tale approccio sia in linea con il quadro normativo esistente e sia stato preventivamente concordato con l'autorità.
Il campo di applicazione degli orientamenti copre, tra l'altro, gli aspetti relativi alla conformità e all'applicazione.
Conformità e applicazione: punti chiave
In base alla regola generale, qualora il fabbricante di dispositivi medici non rispetti i requisiti normativi applicabili, l'autorità è tenuta a intraprendere azioni coercitive.
Qualora il produttore decida di presentare una dichiarazione di conformità per dimostrare la conformità a specifici requisiti normativi, sarà necessario dichiarare la conformità alle norme IEC pertinenti in virtù del seguente programma di prove appropriato. Il sistema di qualità che deve essere sviluppato e attuato dal fabbricante di dispositivi medici dovrebbe affrontare debitamente vari aspetti relativi alla sicurezza dalle radiazioni e alla conformità alle norme applicabili attraverso la verifica e la convalida del progetto. Inoltre, si afferma anche che i risultati di tali prove dovrebbero essere debitamente documentati e che i registri pertinenti dovrebbero essere conservati dal fabbricante come prescritto dai requisiti di conservazione dei registri applicabili da fornire all'autorità su richiesta. Secondo la guida, La FDA considererà un prodotto in violazione degli standard di prestazione del prodotto elettronico se la FDA rileva che il programma di test di un produttore non garantisce l'adeguatezza delle misure di protezione contro le radiazioni pericolose del prodotto elettronico o che non assicura che i prodotti elettronici siano conformi agli standard appropriati.
Come ulteriormente descritto nel documento, quando rilascia una dichiarazione di conformità e dimostra la conformità alle norme pertinenti e ai relativi emendamenti, il produttore dichiara e conferma che sono state stabilite le specifiche di progettazione appropriate in termini di emissione di radiazioni. Qualora il prodotto in questione non sia conforme alle suddette specifiche, si considera difettoso il prodotto elettronico. In tal caso, il prodotto potrebbe essere soggetto a riacquisto, riparazione o sostituzione come richiesto dai requisiti normativi applicabili.
Allo stesso tempo, l'autorità sottolinea inoltre che il presente documento non intende introdurre modifiche all'approccio che l'autorità applica rispetto all'esecuzione della correzione di tali difetti. Come prescritto dalla normativa in materia, i fabbricanti di dispositivi medici, così come gli importatori di dispositivi medici, sono obbligati a notificare all'autorità qualsiasi difetto di radioprotezione identificato, mentre l'autorità, a sua volta, notificherà all'autorità i problemi rilevati. Il regolamento 21 CFR parte 1004 richiede a un produttore di dispositivi medici di intraprendere le azioni necessarie per affrontare il problema identificato, tra cui, tra l'altro, il riacquisto, la riparazione o la sostituzione gratuita di un prodotto interessato in conformità con il piano che deve essere approvato dall'autorità. Il regolamento applicabile autorizza l'autorità a rivedere e approvare o rifiutare i piani di azioni correttive sviluppati dai produttori di dispositivi medici.
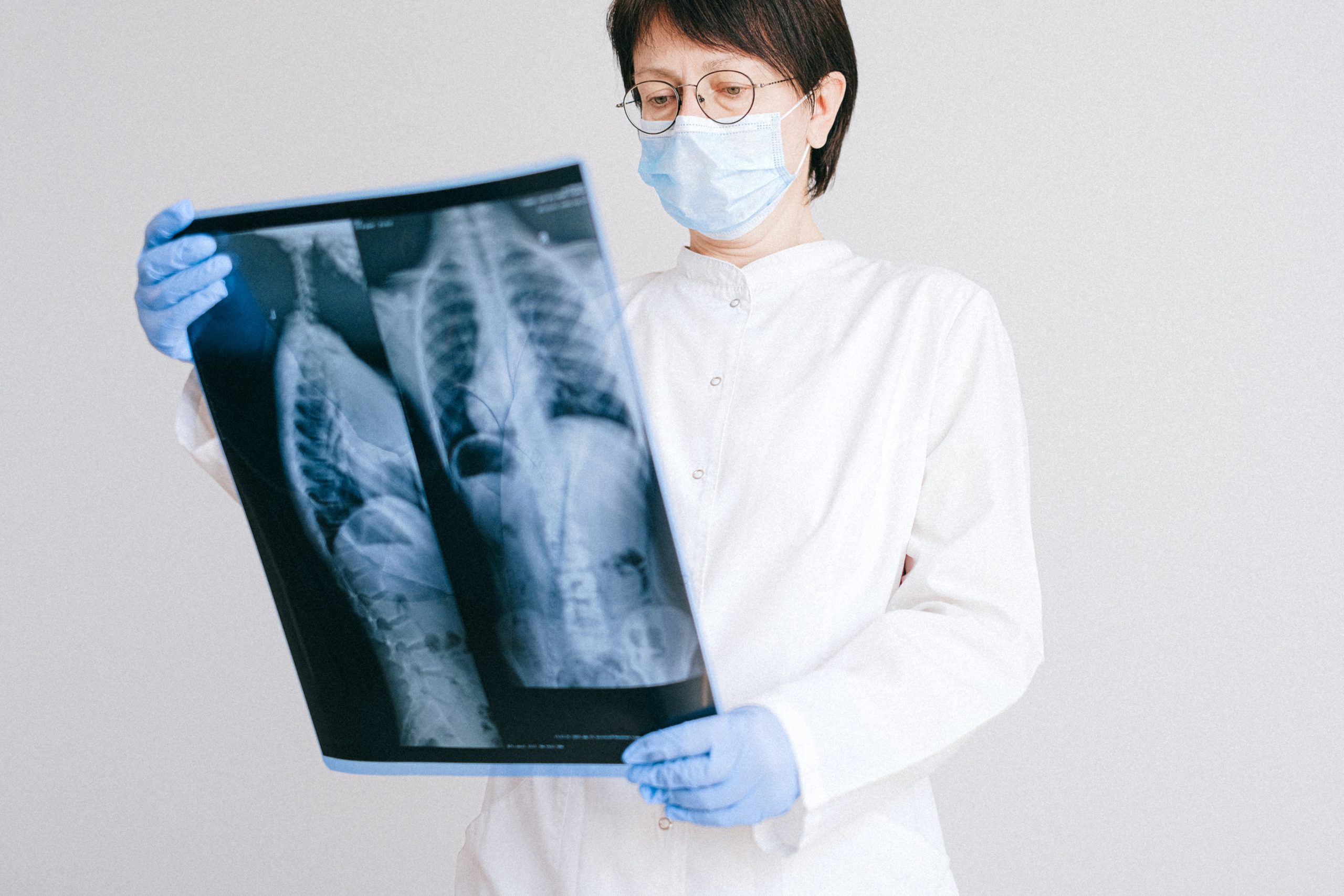
Standard IEC e tipi di dispositivi specifici
Il documento descrive anche gli aspetti relativi all'applicabilità delle norme IEC a specifiche tipologie di dispositivi. In particolare, l'IEC si basa su una struttura a livelli degli standard: standard generali, standard collaterali e standard particolari. Questi tipi di standard devono essere interpretati come segue:
- Lo standard di base (ad es. IEC 60601-1 per le apparecchiature elettromedicali) è chiamato standard generale;
- Gli standard collaterali (ad es. IEC 60601-1-3 per la protezione dalle radiazioni nelle apparecchiature diagnostiche a raggi X) forniscono specifiche generali per la sicurezza applicabili a un sottogruppo di dispositivi coperti dallo standard generale o una caratteristica specifica di tutte le apparecchiature coperte dallo standard standard generale che non è completamente affrontato nello standard generale.
- Norme particolari si applicano a tipi specifici di apparecchiature (ad esempio, IEC 60601-2-43 per i sistemi di fluoroscopia interventistica) e possono sostituire, aggiungere, modificare o rimuovere le condizioni contenute nelle norme generali o collaterali, a seconda del tipo specifico di attrezzature in esame. Questo tipo di standard potrebbe essere utilizzato anche per aggiungere dettagli relativi alla sicurezza e alle prestazioni.
Come ulteriormente spiegato dall'autorità, i riferimenti alle norme applicabili si applicano di norma a tutte le suddette tipologie di norme. È inoltre importante ricordare che in caso di condizioni contrastanti, uno standard particolare prevarrà su quelli collaterali e generali.
In sintesi, la presente guida della FDA fornisce ulteriori chiarimenti sul modo in cui gli standard IEC dovrebbero essere applicati rispetto alle apparecchiature mediche per l'imaging a raggi X. L'ambito di applicazione della guida copre gli aspetti relativi alle azioni di applicazione che l'autorità può intraprendere in risposta alle non conformità identificate e descrive anche i tipi esistenti di standard a cui i fabbricanti di dispositivi medici possono fare riferimento per dimostrare la conformità con la sicurezza e le prestazioni applicabili. relativi requisiti.
Fonte:
Come può aiutare RegDesk?
RegDesk è un sistema olistico di gestione delle informazioni normative che fornisce alle aziende di dispositivi medici e farmaceutiche intelligence normativa per oltre 120 mercati in tutto il mondo. Può aiutarti a preparare e pubblicare applicazioni globali, gestire gli standard, eseguire valutazioni delle modifiche e ottenere avvisi in tempo reale sulle modifiche normative attraverso una piattaforma centralizzata. I nostri clienti hanno anche accesso alla nostra rete di oltre 4000 esperti di conformità in tutto il mondo per ottenere la verifica su questioni critiche. L'espansione globale non è mai stata così semplice.
Vuoi saperne di più sulle nostre soluzioni? Parla oggi con un esperto di RegDesk!
- Distribuzione di contenuti basati su SEO e PR. Ricevi amplificazione oggi.
- Platoblockchain. Web3 Metaverse Intelligence. Conoscenza amplificata. Accedi qui.
- Fonte: https://www.regdesk.co/fda-guidance-on-x-ray-devices-and-iec-standards-enforcement-actinos-and-standard-types/
- :È
- 1
- 8
- a
- Chi siamo
- accesso
- Secondo
- Action
- azioni
- aggiuntivo
- Inoltre
- indirizzo
- indirizzi
- adeguatezza
- amministrazione
- avanzare
- contro
- agenzia
- Tutti
- alternativa
- modifiche
- ed
- applicabile
- applicazioni
- applicato
- APPLICA
- approccio
- opportuno
- approvare
- approvato
- SONO
- articolo
- AS
- aspetti
- valutazioni
- At
- autorità
- base
- basato
- BE
- essendo
- by
- detto
- Materiale
- Custodie
- centralizzata
- il cambiamento
- Modifiche
- caratteristica
- carica
- clienti
- Collaterale
- Aziende
- conformità
- condizioni
- Conflitto
- Prendere in considerazione
- considerazione
- considerato
- potuto
- coperto
- copre
- critico
- decide
- dichiara
- dedicato
- dimostrare
- dimostrando
- descritta
- Design
- dettagli
- sviluppato
- dispositivo
- dispositivi
- scoperto
- documento
- droga
- e
- Elettronico
- elettroni
- emissione
- sottolinea
- applicazione
- garantire
- usate
- sviluppate
- esistente
- espansione
- esperto
- esperti
- ha spiegato
- FAIL
- fda
- trova
- seguito
- i seguenti
- segue
- cibo
- Food and Drug Administration
- Nel
- Contesto
- completamente
- ulteriormente
- Inoltre
- Generale
- globali
- espansione globale
- Avere
- assistenza sanitaria
- Aiuto
- olistica
- HTTPS
- identificato
- Imaging
- implementato
- importante
- imporre
- in
- Compreso
- informazioni
- Intelligence
- introdurre
- coinvolto
- problema
- sicurezza
- emittente
- IT
- SUO
- jpg
- Le
- Sapere
- Legale
- linea
- gestire
- gestione
- sistema di gestione
- Costruttore
- Produttori
- Mercati
- max-width
- medicale
- dispositivo medico
- dispositivi medici
- Scopri di più
- Inoltre
- Natura
- necessaria
- Rete
- New
- obblighi
- ottenere
- of
- on
- minimo
- Altro
- parte
- particolare
- parti
- performance
- Pharma
- piano
- piani
- piattaforma
- Platone
- Platone Data Intelligence
- PlatoneDati
- Preparare
- presenti
- Prodotto
- Prodotti
- Programma
- protezione
- fornire
- purché
- fornisce
- pubblicare
- pubblicato
- qualità
- domanda
- Domande
- Radiazione
- tempo reale
- raccomandazioni
- registrazione
- record
- Riferimenti
- per quanto riguarda
- Regolamento
- normativo
- relazionato
- pertinente
- rimuovere
- riparazione
- sostituire
- richiesta
- necessario
- Requisiti
- richiede
- risposta
- Risultati
- recensioni
- Regola
- norme
- Correre
- Sicurezza
- Suddetto
- stesso
- portata
- dovrebbero
- Un'espansione
- da
- Soluzioni
- fonti
- parlare
- specifico
- Specifiche tecniche
- Standard
- standard
- ha dichiarato
- La struttura
- soggetto
- inviare
- tale
- SOMMARIO
- sistema
- SISTEMI DI TRATTAMENTO
- Fai
- condizioni
- Testing
- che
- I
- loro
- Attraverso
- tempo
- Titolo
- a
- TURNO
- Tipi di
- per
- us
- uso
- generalmente
- convalida
- vario
- Convalida
- VIOLAZIONE
- Modo..
- WELL
- while
- volere
- con
- senza
- In tutto il mondo
- raggi X
- zefiro











