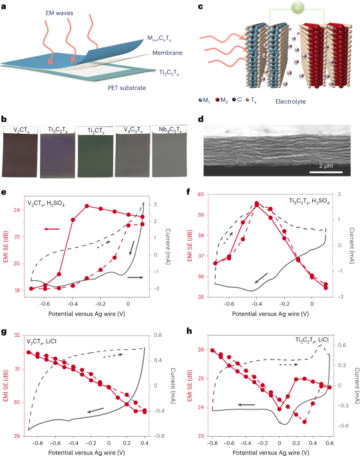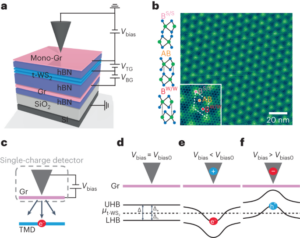
患者样本采集
如前所述,通过擦拭患者的喉咙后部(口咽拭子)收集患者样本26. 这些样本是从具有类似 COVID-19 临床表现的患者身上采集的,并在提取核酸后用 qRT-PCR 进行了检测。 简而言之,收集后,将拭子放入含有裂解缓冲液(4 M 硫氰酸胍、25 mM Tris–HCl、0.5% β-巯基乙醇和 MS2 RNA(200 ng µl - 1; 罗氏))。 轻轻搅动试管以确保裂解缓冲液均匀分布。 之前已经描述了安全步骤,并且是在经过认证的 CL2 实验室中执行的26.
核酸提取
使用基于离心柱的系统提取总核酸,并用于标准化 qRT-PCR 测试26. 内部放大控制 (MS2 (~6 × 104 PFU毫升 - 1) 每 10 ml 裂解缓冲液) 添加到补充裂解缓冲液中 (25 µl 每 10 ml 裂解缓冲液)。 样品在 100 µl 无核酸酶水 (nfH2哦; Invitrogen) 静置 1 分钟,然后以 1× 离心 21,130 分钟g (15,000 rpm) 在台式微量离心机中。 洗脱的样品直接进行 qRT-PCR。 剩余的核酸提取物储存在-80°C,并进一步用于纳米诱饵-纳米孔传感。
SARS-CoV-2 的 qRT-PCR
如前所述进行 SARS-CoV-2 检测26. 每次反应,预混液包含 12.5 µl 2× Luna Universal Probe 一步反应混合物,0.5 µl 20 µM Wu 正向引物(5'-ATGGGTTGGGATTATCCTAAATGTGA-3'),0.5 µl 20 µM Wu 反向引物(5' -GCAGTTGTGGCATCTCCTGATGAG-3'), 0.3 µl 10 µM MGB 探针 3 荧光素 (5'-ATGCTTAGAATTATGGCCTCAC-3'), 0.5 µl 10 µM MS2 RNA 内部对照正向引物, 0.5 µl 10 µM 内部对照反向引物MS2 RNA、0.3 µl 10 µM 内部探针 (MS2 ROX)、1 µl Luna WarmStart RT 酶混合物和 3.9 µl nfH2O. 然后,将 20 µl 主混合物分装到 96 孔板的每个孔中,然后与 5 µl 每种提取物混合。 接受完整提取协议的 MS2 内部提取和扩增控制作为负提取控制包含在 qRT-PCR 板上至少两个孔中。 为了确定 qRT-PCR 过程中的潜在污染,5 µl nfH2O 作为 qRT-PCR 阴性对照。 然后,将 5 微升掺加的 SARS-CoV-2 模板质粒包含在一个孔中作为 qRT-PCR 阳性对照。 将 5 µl 的每个样品添加到其指定的孔中后,用光学透明塑料密封件密封板。 将板以 1× 离心 2,000 分钟g (1,000 rpm) 在 4 °C,然后插入 qRT-PCR 机器(QuantStudio,Thermo Fisher Scientific)并对运行进行参数化。 获得了荧光素 (FAM) 和羧基罗丹明 (ROX) 的信号。 ROX 用于检测内部 MS2 对照,荧光素用于检测 SARS-CoV-2 RNA。 该测定在 2°C 下进行 25 分钟,在 15°C 下进行 50 分钟(对于逆转录酶),在 2°C 下进行 90 分钟,然后进行 45 个循环,95°C 3 秒,然后 60°C 30 秒. 结果通过确认正确的阳性对照(质粒扩增)、所有样品的提取和扩增对照(ROX通道)、阴性对照中无扩增和对照的平均值一致来确定。 SARS-CoV-2 阳性通过荧光素通道中的扩增得到证实,具有适当的 S 形曲线,CT 值≤36。 保持 MS2 和 MGB 探针 3 的 CT 值以跟踪测定的质量和重现性44.
用于纳米诱饵的可编程 RNase H 切割
对于纳米孔传感,SARS-CoV-2 RNA 对照、核酸提取物(患者样本)或 MS2 病毒 RNA 被进一步用于纳米诱饵检测。 首先,我们将引导寡核苷酸与样品混合并将其加热至 70 °C 5 分钟。 添加 RNase H(每毫升 5,000 单位;NEB),混合并在 20 °C 下加热 37 分钟,以允许该酶切割 DNA 中的 RNA:RNA 杂交体,从而有效释放目标 RNA。 通过在 65°C 下孵育 10 分钟,使 RNase H 热失活。 使用 NUPACK 软件验证引导寡核苷酸不会形成分子内结构、同二聚体或异二聚体45. 对于缺少目标的测量,使用了相同的协议,包括引导寡核苷酸。 控制测量显示没有位移,因此,我们可以从引导寡核苷酸中排除任何实质性的交叉结合。
纳米诱饵的病毒靶序列特性
选择目标长度、立足点长度和 GC 含量以确保最佳杂交21. 对于 DNA 纳米诱饵设计,目标序列被选择在病毒基因组的保守区域,并且具有 40-60% 的 GC 含量以形成稳定的 20 bp 双链体。 立足点长度选择为 6 nt 长,GC 含量为 40–60%。 我们使用 NUPACK 软件(网络应用程序 2020)测试了所有序列是否存在潜在的不良高度稳定的分子内相互作用或同型二聚体45. 然后,我们在每个实验中使用的多个站点之间进行了交叉反应检查45.
纳米诱饵DNA花的制备
我们设计了一个 DNA 花作为从患者样本中检测 SARS-CoV-2 RNA 的另一个标签。 分别制备了针对每个 SARS-CoV-2 靶标的三种 DNA 花(七向连接,7WJa、7WJb 和 7WJc)。 以7WJc为例,4 μM DNA链J1、J2、J3和J4c(附表 1) 在 TM 缓冲液(10 mM Tris–HCl、10 mM MgCl2, pH 8.0) 并加热至 90 °C 5 分钟,然后冷却至 65 °C 15 分钟,45 °C 15 分钟,37 °C 20 分钟,25 °C 20 分钟,最后至 4 °C C 20 分钟。 链 J4c 被 J4b 取代以制备 7WJb。 对于 7WJa,为了避免在纳米诱饵的 43 位点发生自折叠,J1、J2、J3 J4a 和 C43 在退火前混合在一起。
DNA纳米诱饵的自组装
通过将线性化单链 M13 DNA(M13mp18,7,249 nt,Guild Biosciences,100 nM)与短互补寡核苷酸混合来组装 DNA 纳米诱饵12 (其中一些包含参考结构和捕获链)并通过添加部分互补链,这些链被 3'-生物素化以用于立足点介导的链置换反应。 线性化的 M13 DNA(长度为 7,228 nt)由寡核苷酸补充,从而产生带切口的双链纳米诱饵,其两端有四个脱氧胸苷突出端,可防止多聚化12. 该混合物包含 20 nM 线性化 M13 DNA、60 nM 寡核苷酸(超过 M13 DNA 的三倍)、浓度为 3 nM 的 180'-生物素化链、10 mM MgCl2 和 1× TE(10 mM Tris–HCl,1 mM EDTA,pH 8.0)。 将其通过移液器混合并在加热至 70°C 30 秒之前旋转并在 45 分钟内冷却至环境温度。 使用具有 0.5 kDa 截止值和洗涤缓冲液(100 mM Tris-HCl pH 10.0, 8.0 mM MgCl2). 如果 DNA 花被用作标签,则携带它的部分互补链在 10 mM MgCl 中孵育2 在环境温度下持续 2 小时,随后,如上所述进行 Amicon 过滤。 纳米诱饵设计的不对称性允许明确识别结合位点。 纳米诱饵在 4-10 °C 的 0.5 mM MgCl 中储存直至用于进一步实验2, 10.0 mM Tris–HCl, pH 8.0。 在每次测量之前,通过纳米孔读数检查纳米诱饵设计。
DNA 纳米诱饵的纳米孔读数
纳米诱饵与样品(核酸提取物或纯化的病毒靶标,过量十倍)在 10 mM MgCl 中混合2 和 100 毫米氯化钠。 将混合物 (5 μl) 在室温下孵育(约 10 分钟),直至准备好用于纳米孔测量。 目标序列组成及其物理特性的差异可能导致杂交的可变性,从而导致传感位点的置换效率21. 我们使用了 htRNA (100 ng μl - 1; Invitrogen) 作为指示的背景,以表明不存在由人类天然 RNA 诱导的非特异性信号。 对于纳米孔测量,将样品稀释至 <0.5 nM 纳米诱饵(用于纯化的病毒靶标)或将 4.7 μl RNase-H 切割患者样品与 0.3 μl 单价链霉亲和素 (SAe1D3) 混合18 (1 μM)、5 μl LiCl (4.0 M) 和 5.0 μl LiCl (8.0 M)。 我们制造了 14 ± 3 nm(平均值 ± 标准偏差)的纳米孔12 通过激光辅助拉拔器 P-0.5(萨特仪器)使用外径为 0.2 毫米、内径为 2000 毫米的石英玻璃毛细管(萨特仪器)。 将混合物移入纳米孔聚二甲基硅氧烷芯片中,所有测量均在 600 mV 的恒定电压下进行。 纳米孔测量细节显示在补充表中 30.
实时纳米孔数据分析
补充部分详细解释了纳米孔数据分析 14. 简而言之,从原始离子电流迹线中过滤出纳米诱饵事件,然后确定检测区域,并提取尖峰在每个特定位点存在的信息。 绘制的置换效率被计算为测量的置换效率减去每个站点的无目标控制(三个纳米孔记录中的每一个的 50 个纳米诱饵事件),除非另有说明:
$$begin{array}{l}{mathrm{Displacement}},{mathrm{efficiency}} =frac{1}{3}mathop {sum}limits_{n = 1}^3 left{ {1 -frac{1 }{{50}}mathop {sum}limits_{n = 1}^{50} {left[ {fleft( n right) = left( {frac{{1,,mathrm{peak}}}{{0,, {mathrm{no}},{mathrm{peak}}}}} right)} right]_{{{{mathrm{target}}}}}} } right}\ – frac{1}{3}mathop {sum }limits_{n = 1}^3 {left{ {1 – frac{1}{{50}}mathop {sum }limits_{n = 1}^{50} left[ {fleft( n right) = left( { frac{{1,,mathrm{peak}}}{{0,,mathrm{no}},{mathrm{peak}}}} right)} right]_{{{mathrm{no}}}},{ {{mathrm{target}}}}}} right}} end{array}.$$
我们验证了卷积神经网络 QuipuNet27 能够按照所描述的程序对纳米孔数据进行实时分析。 之前,我们证明了通过大约 99 个事件,我们对我们设计的 DNA 结构的阳性检测有 XNUMX% 的置信度46.
原子力显微镜成像
纳米诱饵的 AFM(Nanosurf Mobile S)成像是在空气中以非接触模式进行的。 纳米诱饵结构被稀释至 1 ng μl - 1 在 1 mM MgCl2 将 10 μl 加入新鲜切割的云母中,孵育 1 分钟,用过滤的 Milli-Q 水冲洗,然后用氮气吹干。 扫描前,使用双面胶带将云母板固定到 AFM 样品台上。 使用 Gwyddion(版本 2.60)进行图像可视化和分析。
统计分析
对于所有测量,计算了置换效率的 99.9% 置信区间。 使用双侧 Student's 测试没有和有目标的两个站点之间的统计显着性 t检验。
报告摘要
有关研究设计的更多信息,请参阅 自然投资组合报告摘要 链接到这篇文章。
- SEO 支持的内容和 PR 分发。 今天得到放大。
- 柏拉图区块链。 Web3 元宇宙智能。 知识放大。 访问这里。
- Sumber: https://www.nature.com/articles/s41565-022-01287-x
- 000
- 1
- 10
- 100
- 11
- 110
- 2014
- 2016
- 2017
- 2018
- 2020
- 2021
- 7
- 70
- 9
- a
- 以上
- 缺席
- 学者
- 后天
- 添加
- 后
- 加拿大航空
- 所有类型
- 允许
- 环境
- 分析
- 锚
- 和
- 另一个
- 应用领域
- 的途径
- 适当
- 围绕
- 刊文
- 组装
- 可使用
- 背部
- 背景
- before
- 钟
- 之间
- 捆绑
- 吹
- BP
- 简要地
- 缓冲
- 计算
- 能力
- 捕获
- 携带
- 认证
- 渠道
- 特点
- 查
- 陈
- 芯片
- 清除
- 临床资料
- 采集
- 结合
- 补充
- 浓度
- 信心
- CONFIRMED
- 一贯
- 常数
- 内容
- 控制
- 控制
- 卷积神经网络
- 创造
- 电流
- 曲线
- 切
- 切割
- 周期
- data
- 数据分析
- 证明
- 描述
- 设计
- 设计
- 设计
- 细节
- 详情
- 检测
- 确定
- 决心
- 偏差
- 差异
- 数字
- 直接
- 分配
- 的DNA
- 向下
- 每
- 只
- 效率
- 效率
- 工程师
- 确保
- 错误
- 醚(ETH)
- 甚至
- 事件
- 例子
- 解释
- 提取
- 萃取
- 提取物
- 过滤器
- 终于
- (名字)
- 花
- 其次
- 以下
- 申请
- 向前
- 止
- ,
- 进一步
- 基因组
- 玻璃
- 指南
- 高度
- HTTPS
- 人
- 杂交种
- 鉴定
- 图片
- 同步成像
- 履行
- in
- 包括
- 包含
- 孵化
- 孵化
- 信息
- 仪器
- 互动
- 内部
- 离子的
- IT
- KDA
- 标签
- 铅
- 长度
- 友情链接
- 链接
- 长
- 卢纳
- 机
- 主
- 材料
- 测量
- 云母
- 可能
- 最低限度
- 杂
- 搅和
- 混合物
- ML
- 联络号码
- 时尚
- MOL
- 多
- 纳米
- 纳米孔
- 本地人
- 自然
- 负
- 网络
- 神经
- 神经网络
- 打开
- 最佳
- 除此以外
- 配对
- 病人
- 患者
- 性能
- 的
- 图片
- 塑料
- 柏拉图
- 柏拉图数据智能
- 柏拉图数据
- 个人档案
- 积极
- 阳性
- 潜力
- Prepare
- 准备
- 存在
- 防止
- 先前
- 底漆
- 探测器
- 过程
- 蛋白质
- 协议
- 质量
- 原
- 达到
- 反应
- 实时的
- 地区
- 地区
- 发布
- 其余
- 去除
- 报告
- 研究
- 成果
- 反转
- RNA
- 罗氏
- Room
- rt
- 定位、竞价/采购和分析/优化数字媒体采购,但算法只不过是解决问题的操作和规则。
- 运行
- 实现安全
- 同
- SARS-COV-2
- 扫描
- 部分
- 选
- 序列
- 短
- 显示
- 如图
- 信号
- 意义
- 同时
- 单
- 网站
- 网站
- 小
- 软件
- 一些
- 具体的
- 纺
- 稳定
- 阶段
- 站
- 标准
- 说
- 统计
- 步骤
- 存储
- 链
- 后来
- 大量
- 产品
- 表
- 服用
- 目标
- 目标
- 模板
- 十
- 从而
- 三
- 时
- TM
- 至
- 一起
- 合计
- 跟踪时
- 超级
- 下
- 单位
- 普遍
- 验证
- 折扣值
- 价值观
- 专利
- 版本
- 通过
- 病毒
- 可视化
- 电压
- W
- 水
- 卷筒纸
- Web应用程序
- 井
- 这
- 也完全不需要
- wu
- 和风网