DNA origami การพับและการทำให้บริสุทธิ์
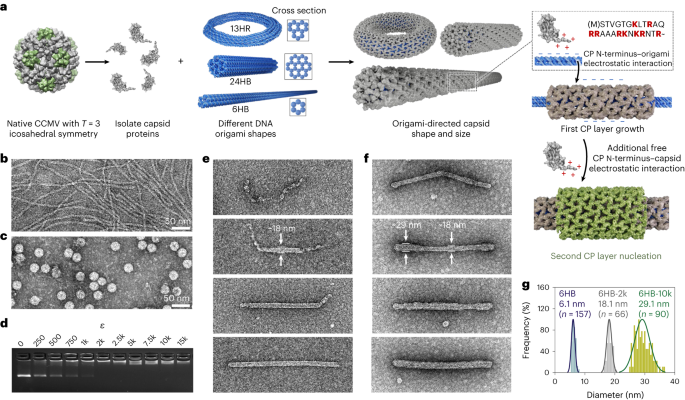
โครงสร้าง DNA origami (6HB, 24HB, 60HB, 13HR และ nanocapsule) ถูกพับในปฏิกิริยาแบบหม้อเดียวโดยค่อยๆ ลดอุณหภูมิโดยใช้ระบบ PCR Proflex 3 × 32 หลุม (เทอร์โมฟิชเชอร์) เส้นนั่งร้าน (ตัวแปร p7249, p8064 และ p7560 ของ M13mp18 แบบเส้นเดี่ยว) ถูกซื้อจาก Tilibit Nanosystems และเส้นหลักจาก Integrated DNA Technologies เพื่อให้แน่ใจว่าอัตราการพับของ DNA origami สูง จึงมีการใช้เงื่อนไขที่ได้รับการปรับปรุงเฉพาะโครงสร้างที่เกี่ยวข้องกับทั้งขั้นตอนการหลอมและตัวเลือกบัฟเฟอร์ ('บัฟเฟอร์การพับ', FOB) (หมายเหตุเสริม) 20).
การแลกเปลี่ยนบัฟเฟอร์สำหรับ DNA origami
โครงสร้าง DNA origami บริสุทธิ์ถูกถ่ายโอนไปยังบัฟเฟอร์ 6.5 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) เสริมด้วย NaOH 2 mM (HEPES-NaOH, pH 6.5) ก่อนทำให้เกิดซ้อนด้วย CCMV CPs การแลกเปลี่ยนบัฟเฟอร์ถูกดำเนินการโดยการกรองแบบหมุน59 ใช้ตัวกรองแบบแรงเหวี่ยงแบบตัดน้ำหนักโมเลกุล (MWCO) 100 kDa (Amicon) ซึ่งถูกล้างก่อนใช้งานโดยการปั่นแยกด้วยบัฟเฟอร์ HEPES-NaOH 400 μl เป็นเวลา 5 นาทีที่ 14,000g. ต่อจากนั้น ปริมาตรที่เท่ากันของสารละลาย DNA origami และบัฟเฟอร์ HEPES-NaOH ถูกเติมเข้าไปในอุปกรณ์กรอง และการหมุนเหวี่ยงดำเนินต่อไปเป็นเวลา 10 นาทีที่ 6,000g. จากนั้นจึงเติมปริมาตร HEPES-NaOH เท่ากับ 2.09 × ปริมาตรเริ่มต้นของสารละลายโอริกามิ และขั้นตอนการปั่นเหวี่ยงซ้ำ เก็บตัวอย่างโดยการกลับตัวกรองและปั่นแยกเป็นเวลา 2.5 นาทีที่ 1,000g.
การแยก CCMV CPs
CP ถูกแยกออกจาก CCMV ที่ไม่บุบสลาย (สำหรับการเตรียมไวรัส ดูหมายเหตุเพิ่มเติม 21). โดยสรุป อนุภาคไวรัสถูกฟอกข้ามคืนกับ Tris–HCl 50 mM, CaCl 500 mM2 บัฟเฟอร์ pH 7.5 เสริมด้วยไดไทโอไทรทอล (DTT) 1 มิลลิโมลาร์โดยใช้ถ้วยไตเทียมขนาดเล็ก Slize-A-Lyzer (3.5 kDa MWCO, Thermo Scientific) RNA ถูกอัดเป็นก้อนในขั้นตอนการปั่นแยกที่ 4 °C โดยใช้ 21,100g เป็นเวลา 6 ชั่วโมง และส่วนลอยเหนือตะกอนที่คืนสภาพกลับคืนมาจะถูกฟอกข้ามคืนกับ 'บัฟเฟอร์ที่สะอาด' ที่มี Tris–HCl 50 mM, NaCl 150 mM ที่ pH 7.5 เสริมด้วย DTT 1 mM (ดัดแปลงจากการอ้างอิง 60). ความเข้มข้นของโปรตีนถูกกำหนดตามการดูดกลืนแสงที่ 280 นาโนเมตร (สัมประสิทธิ์การสูญพันธุ์ 23,590 M-1 cm-1) โดยใช้เครื่องสเปกโตรโฟโตมิเตอร์ไมโครเพลท BioTek Eon (ตัวอย่าง 2 μl, เพลต Take3)
อายุ
AGE ถูกนำมาใช้เพื่อศึกษาปฏิสัมพันธ์ที่มีผลผูกพันระหว่างโปรตีนและโครงสร้าง origami โดยการตรวจสอบการเปลี่ยนแปลงในการเคลื่อนที่ด้วยไฟฟ้า นอกจากนี้ ความสมบูรณ์ของโครงสร้างโอริกามิหลังจากการพับและการทำให้บริสุทธิ์ และในระหว่างการย่อย DNase I ได้รับการวิเคราะห์โดยเจลอิเล็กโทรโฟเรซิส ด้วยเหตุนี้ ตัวอย่าง (ปริมาตรตั้งแต่ 10 ถึง 32 ไมโครลิตร) ที่เสริมด้วยสีย้อมเจล 6 เท่า (ซูโครส 40% ที่ไม่มีสีย้อมสำหรับตัวอย่างจากการศึกษาการย่อยอาหาร) จึงถูกนำไปใช้ในเจลอะกาโรส 2% (w/v) (1 × ทริส –อะซิเตต–เอทิลีนไดเอมีนเตตร้าอะซิติกแอซิด (TAE) บัฟเฟอร์, 11 มิลลิโมลาร์ MgCl2) เป็นเวลา 45 นาทีที่ 90 V ในบัฟเฟอร์ 1 × TAE เสริมด้วย MgCl 11 mM2. สำหรับการย้อมสี เอทิเดียมโบรไมด์ (EtBr) ที่ความเข้มข้นสุดท้ายคือ 0.46 ไมโครกรัม มล.-1 ถูกนำมาใช้และมองเห็น DNA ภายใต้แสงอัลตราไวโอเลตโดยใช้ระบบ GelDoc XR+ (Bio-Rad)
ความซับซ้อนของ DNA origami และ CCMV CPs
ความซับซ้อนระหว่าง CPs และ DNA origami ดำเนินการที่ความเข้มข้น origami สุดท้ายที่ 4 nM (ตัวอย่าง 10 μl) มีการเติมโอริกามิในอัตราส่วนปริมาตร 1:1 ต่อสารละลายโปรตีนที่เจือจางใน 'บัฟเฟอร์ที่สะอาด' ขึ้นอยู่กับปริมาณโปรตีนส่วนเกินที่ต้องการ εซึ่งอธิบายอัตราส่วนฟันกรามระหว่าง CP ต่อ DNA origami สารละลายโปรตีนตั้งแต่ 0 ถึง 60 μM (สอดคล้องกับ ε = 0–15k) เตรียมไว้แล้ว ความเข้มข้นของ NaCl ถูกปรับเป็น 150 มิลลิโมลาร์ ส่งผลให้บัฟเฟอร์เชิงซ้อนประกอบด้วย HEPES-NaOH 3.25 มิลลิโมลาร์, Tris–HCl 25 มิลลิโมลาร์, NaCl 150 มิลลิโมลาร์ และ DTT 0.5 มิลลิโมลาร์ ทำการเกิดภาวะเชิงซ้อนที่ 4 °C เป็นเวลาอย่างน้อย 1 ชั่วโมง และวิเคราะห์ในเวลาต่อมาโดยใช้ AGE และ TEM
DNase I ตรวจการย่อยอาหาร
เพื่อศึกษาผลการป้องกันของการเคลือบ CP ต่อการเสื่อมสภาพของโครงสร้างโอริกามิโดย DNase I, สต็อก DNase I 2 μl (ตั้งแต่ 0 ถึง 500 KU ml-1) ถูกเติมลงในตัวอย่าง 16 ไมโครลิตร นอกจากนี้ CaCl2 และ MgCl2 ปรับความเข้มข้นส่งผลให้ปริมาตรปฏิกิริยาสุดท้าย 20 μl ประกอบด้วย DNA origami 3.2 nM, HEPES-NaOH 2.6 mM, Tris – HCl 20 mM, NaCl 120 mM, DTT 0.4 mM, CaCl 1 mM2 และ MgCl 5 มิลลิโมลาร์2. ตัวอย่างถูกบ่มที่ 37 °C เป็นเวลา 15 นาที (6HB) และ 60 นาที (24HB) ก่อนที่จะวิเคราะห์ผลลัพธ์โดย AGE ตัวอย่างที่ซับซ้อนด้วย CPs จะถูกแยกชิ้นส่วนโดยใช้เกลือเฮปารินโซเดียมเป็นสารยึดเกาะที่แข่งขันได้ (ความเข้มข้นสุดท้ายที่ 1.5 μM สำหรับ 6HB-2k และ 24HB-2.5k และ 82 μM สำหรับ 6HB-10k และ 24HB-10k; เสริม บันทึก 14).
การพับกระดาษ RNA – DNA และการทำให้บริสุทธิ์
สำหรับ RNA – DNA origami ไฮบริด (RNA-6HB) จะใช้ EGFP mRNA (CleanCap EGFP mRNA, TriLink Bio Technologies, L-7601) เป็นโครง ในปฏิกิริยาแบบหม้อเดียว โครง mRNA ที่มีความยาว 996 nt ได้รับการอบอ่อนด้วยความร้อนด้วยลวดเย็บกระดาษ 29 เส้น (ซื้อจาก Integrated DNA Technologies ดูหมายเหตุเพิ่มเติม 22) ลงในโครงสร้าง 6HB แบบสั้นโดยใช้ระบบ PCR Proflex 3 × 32 หลุม (เทอร์โมฟิชเชอร์) โครงสร้างได้รับการออกแบบให้มีครอสโอเวอร์นั่งร้านสองตัวและมีระยะพิทช์เกลียวที่ 11 bp ต่อเทิร์น สำหรับปฏิกิริยาการพับ mRNA และลวดเย็บกระดาษถูกเจือจางเป็น 1 × FOB ที่มี 1 × TAE pH 8.4, 5 mM MgCl2 และ NaCl 1 มิลลิโมลาร์ จนถึงความเข้มข้นสุดท้ายที่ 50 นาโนโมลาร์ และ 500 นาโนโมลาร์ ตามลำดับ ของผสมปฏิกิริยาถูกบ่มที่ 55 °C เป็นเวลา 15 นาที61 และทำให้เย็นลงโดยวางบนน้ำแข็งอย่างน้อย 10 นาที ก่อนเก็บรักษาที่อุณหภูมิ 4 °C เพื่อตรวจสอบความถูกต้องของการพับ มีการแลกเปลี่ยนลวดเย็บกระดาษสี่เส้นกับลวดเย็บกระดาษที่มีส่วนยื่น 3 ′ (ติดป้ายกำกับด้วย F ตารางเสริม 2). สายยึดที่มีฟลูออโรฟอร์ (ATTO590, Integrated DNA Technologies) ซึ่งถูกเติมลงในส่วนผสมพับในส่วนที่เกิน 10 เท่าต่อจุดยึด จากนั้นสามารถรวมเข้ากับโครงสร้างได้โดยการไฮบริดไดเซชันกับส่วนที่ยื่นของลวดเย็บ
โครงสร้างที่พับแล้วถูกทำให้บริสุทธิ์จากลวดเย็บกระดาษส่วนเกินโดยการกรองแบบหมุน ด้วยเหตุนี้ ตัวกรอง (100 kDa MWCO, Amicon) จึงถูกล้างด้วย 400 μl ของ 1 × FOB โดยการหมุนเหวี่ยงที่ 14,000g เป็นเวลา 5 นาที ตามด้วยการเติม 40 μl RNA-6HB สองครั้งพร้อมกับ 40 μl ของ 1 × FOB หลังจากปั่นแยกขั้นตอนที่ 6,000g เป็นเวลา 10 นาที เติม 80 μl ของ 1 × FOB และการหมุนเหวี่ยงดำเนินต่อไป (6,000g, 10 นาที) ขั้นตอนการล้างนี้ทำซ้ำทั้งหมดสามครั้งก่อนที่จะนำตัวอย่างกลับคืนมาโดยการกลับตัวกรองลงในท่อที่สะอาด (1,000g, 2.5 นาที) กำหนดความเข้มข้นโดยการวัดค่าการดูดกลืนแสงที่ 260 นาโนเมตร (ค่าสัมประสิทธิ์การสูญพันธุ์ 1.29 × 107 M-1 cm-1) และการพับที่ประสบความสำเร็จถูกกำหนดโดยเจล AGE (3.5 % (w/v) การสร้างภาพภายใต้แสงอัลตราไวโอเลต (ช่อง EtBr) และแสงสีแดง (ช่อง A647) ระบบ ChemiDoc MP, Bio-Rad), AFM และ TEM
ความซับซ้อนของ RNA-6HB origami และ CCMV CPs
สำหรับการทำให้ซับซ้อนนั้น RNA-6HB origami บริสุทธิ์ใน 1 × FOB ถูกผสมกับ CCMV capsids ใน 'บัฟเฟอร์ที่สะอาด' ในอัตราส่วน 1: 1 ที่ความเข้มข้น origami ไฮบริดสุดท้ายที่ 7.5 nM ซึ่งส่งผลให้เกิดบัฟเฟอร์เชิงซ้อนที่ประกอบด้วยทริส 45 มิลลิโมลาร์, NaCl 75.5 มิลลิโมลาร์, กรดอะซิติก 10 มิลลิโมลาร์, MgCl 2.5 มิลลิโมลาร์2, 0.5 มม. DTT และ 0.5 มม. EDTA ตัวอย่างถูกบ่มที่ 4 °C เป็นเวลาอย่างน้อย 1 ชั่วโมงก่อนการวิเคราะห์ด้วย AGE และ TEM
ความซับซ้อนของ DNA origami และ NoV CPs
NoVLP ถูกจัดเตรียมตามที่รายงานโดยแลมพิเนน และคณะ62 และเก็บไว้ในน้ำเกลือบัฟเฟอร์ฟอสเฟต 1 × (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 และ 1.8 mM KH2PO4, pH 7.4); อย่างไรก็ตาม ที่นี่ SpyTag003 (อ้างอิง 63) ถูกหลอมรวมกับปลาย C ของ VP1 จากสายพันธุ์ NoV Hu/GII.4/Sydney/NSW0514/2012/AU อนุภาคถูกควบคุมคุณภาพด้วยการกระเจิงแสงแบบไดนามิกสำหรับการก่อตัวของอนุภาค โซเดียมโดเดซิลซัลเฟตโพลีอะคริลาไมด์เจลอิเล็กโตรโฟรีซิสเพื่อความบริสุทธิ์ของโปรตีน และตรวจวัด dsDNA ที่เหลือ สำหรับการสร้างความซับซ้อนด้วย DNA origami นั้น DNA origami ปรากฏอยู่ในตัวอย่างในระหว่างการถอดชิ้นส่วนและการประกอบ VLP ใหม่ ด้วยเหตุนี้ โครงสร้างการพับกระดาษจึงถูกถ่ายโอนไปยังน้ำปราศจากไอออนโดยใช้การกรองแบบหมุน (ตามที่อธิบายไว้ข้างต้น) การพับกระดาษ DNA ถูกผสมกับ NoVLP ที่ความเข้มข้นต่างกันในอัตราส่วน 1:4 (ปริมาตร/ปริมาตร) ซึ่งส่งผลให้ความเข้มข้นของการพับกระดาษขั้นสุดท้ายอยู่ที่ 6 นาโนโมลาร์ (ตัวอย่าง 30 ไมโครลิตร) ตัวอย่างถูกถ่ายโอนไปยังถ้วยฟอกไต MWCO ขนาด 3.5 kDa (Slize-A-Lyzer, Thermo Scientific) และฟอกไตข้ามคืนที่ 4 °C เทียบกับ Tris–HCl 50 มิลลิโมลาร์, pH 8.9 สำหรับการประกอบซ้ำ ในขั้นตอนที่สอง ตัวอย่างถูกฟอกข้ามคืนที่ 4 °C เทียบกับบัฟเฟอร์โซเดียมฟอสเฟต 100 มิลลิโมลาร์ pH 6.0 ในทำนองเดียวกันตามที่รายงานโดย White และคณะ64 การเกิดภาวะเชิงซ้อนระหว่างการแยกชิ้นส่วนและการประกอบ NoVLP ถูกวิเคราะห์โดย AGE และ TEM
ความซับซ้อนของ DNA origami และ SV40 CPs
SV40 major CP VP1 (abcam, ab74565) ถูกถอดประกอบและประกอบกลับเข้าไปใหม่ (ดัดแปลงจากการอ้างอิง XNUMX) 50) โดยการฟอก VLP ที่ประกอบใน PBS เทียบกับ 20 มิลลิโมลาร์ ทริส, 2 มิลลิโมลาร์ DTT, EDTA 5 มิลลิโมลาร์ และ NaCl 50 มิลลิโมลาร์, pH 8.9 เป็นเวลา 2 ชั่วโมงที่ 4 °C (3.5 กิโลดาลตัน MWCO, Slize-A-Lyzer, Thermo Scientific) หลังจากนั้น ซึ่งความเข้มข้น EDTA ลดลงโดยขั้นตอนการล้างไตเพิ่มเติมที่ 4 °C เป็นเวลา 2 ชั่วโมงเทียบกับ 20 มิลลิโมลาร์ Tris, 2 มิลลิโมลาร์ DTT, EDTA 2 มิลลิโมลาร์ และ NaCl 50 มิลลิโมลาร์, pH 8.9 ความเข้มข้นถูกกำหนดตามการดูดกลืนแสงที่ 280 นาโนเมตร (สัมประสิทธิ์การสูญเสีย VP1, 32,890 M-1 cm-1). DNA origami ถูกถ่ายโอนไปยังบัฟเฟอร์ HEPES 100 มิลลิโมลาร์ pH 7.2 เสริมด้วย NaCl 125 มิลลิโมลาร์โดยการกรองแบบหมุน (ตามที่อธิบายไว้ข้างต้น) โปรตีนถูกผสมกับ DNA origami ในอัตราส่วน 1: 1 (v/v) เพื่อให้ได้ความเข้มข้นสุดท้ายที่ 0–20 μM และ 2 nM ตามลำดับ และตัวอย่างถูกบ่มเป็นเวลา 24 ชั่วโมงที่อุณหภูมิห้องก่อนการวิเคราะห์โดยใช้ AGE และ TEM .
ความซับซ้อนของ DNA origami และ MPyV CPs
สำหรับความซับซ้อนของแคปโซเมอร์ VP1 (สำหรับการแสดงออกและการทำให้บริสุทธิ์แบบรีคอมบิแนนท์ ดูหมายเหตุเพิ่มเติม 23) และ DNA origami โครงสร้างการพับกระดาษถูกถ่ายโอนครั้งแรกไปยัง Tris buffer 40 mM, pH 8.0, เสริมด้วยกรดอะซิติก 20 mM, EDTA 2 mM และ MgCl 12 mM2 โดยใช้การกรองแบบหมุน (ดูด้านบน) ขึ้นอยู่กับปริมาณโปรตีนส่วนเกินที่ต้องการ εแคปโซเมอร์ถูกเจือจางไปเป็น 'บัฟเฟอร์สำหรับการจัดเก็บ' ซึ่งมีทริส 40 มิลลิโมลาร์, NaCl 200 มิลลิโมลาร์, EDTA 1 มิลลิโมลาร์, กลีเซอรอล 5% (ปริมาตร/ปริมาตร) และ DTT 5 มิลลิโมลาร์, pH 8.0 สำหรับการเกิดภาวะเชิงซ้อน VP1 capsomers ถูกเจือจางในอัตราส่วน 1:20 ในสารละลาย origami ส่งผลให้ความเข้มข้นของ origami สุดท้ายอยู่ที่ 0.75 nM (ตัวอย่าง 30 μl) และบัฟเฟอร์เชิงซ้อนที่ประกอบด้วย Tris 40 mM, กรดอะซิติก 19 mM, 1.95 mM EDTA, MgCl 11.4 มิลลิโมลาร์2, NaCl 10 มิลลิโมลาร์, กลีเซอรอล 0.25% (ปริมาตร/ปริมาตร) และ 0.25 มิลลิโมลาร์ DTT, pH 8 ปฏิกิริยาการเกิดภาวะเชิงซ้อนถูกบ่มที่ 4 °C ข้ามคืนก่อนการวิเคราะห์ด้วย AGE และ TEM
AFM
หยด 20 μlของสารละลาย origami 10 nM RNA-6HB (MgCl2 ความเข้มข้นที่ปรับเป็น 12.5 mM) ถูกวางบนพื้นผิวไมกาที่แยกใหม่ (Electron Microscopy Sciences) เป็นเวลา 1 นาที ตามด้วยขั้นตอนการล้างสามขั้นตอนการด้วยน้ำปราศจากไอออน 100 μl ซึ่งถูกลบออกทันที ตัวอย่างถูกทำให้แห้งภายใต้กระแสไนโตรเจนที่สม่ำเสมอ และถ่ายภาพทันทีหลังจากการเตรียมตัวอย่าง ภาพ AFM ได้รับในอากาศโดยใช้ ScanAsyst ในโหมดอากาศร่วมกับหัววัด ScanAsyst-Air (Bruker) บน Dimension Icon AFM (Bruker) การประมวลผลภาพดำเนินการใน NanoScope Analysis v.1.90 (Bruker)
TEM
ตัวอย่าง origami DNA ธรรมดา (4 นาโนโมลาร์) เตรียมโดยการบ่มหยดขนาด 3 μl เป็นเวลา 3 นาทีบนพลาสมาที่ทำความสะอาด (แฟลชพลาสมาออกซิเจน 20 วินาที, Gatan Solarus) กริดทองแดงเคลือบคาร์บอน Formvar (FCF400Cu, Electron Microscopy Sciences) ซึ่งก็คือ ต่อมาซับกับกระดาษกรองและเปื้อนสีลบ สำหรับตัวอย่างที่ซับซ้อน CCMV-CP (โอริกามิ DNA 4 นาโนเมตร) หยดขนาด 3 ไมโครลิตรถูกฝากไว้บนตารางเป็นเวลา 1.5 นาที หลังจากซับกับกระดาษกรองแล้ว กริดจะถูกจุ่มลงในหยดบัฟเฟอร์เชิงซ้อนขนาด 10 ไมโครลิตร (HEPES-NaOH 3.25 mM, Tris–HCl 25 mM, NaCl 150 mM, DTT 0.5 mM) เป็นเวลา 5 วินาที สำหรับตัวอย่างที่มีความเข้มข้นของ DNA origami ≤2 nM (ตัวอย่างเช่น การสร้างสารเชิงซ้อนด้วย SV40, MPyV) และสำหรับตัวอย่างที่มี RNA-6HB (ความเข้มข้นของ origami 7.5 nM) ขนาดหยดจะเพิ่มขึ้นเป็น 5 μl และเวลาฟักตัวขยายเป็น 5 นาที . การย้อมสีเชิงลบ65 ดำเนินการโดยการจุ่มกริดในหยด 5 ไมโครลิตรของสารละลายฟอร์เมตยูรานิลที่มีน้ำ 2% (w/v) (เสริมด้วย NaOH 25 มิลลิโมลาร์สำหรับการปรับ pH) ซึ่งถูกลบออกทันที ขั้นตอนนี้ตามด้วยการแช่ในหยดขนาด 20 ไมโครลิตร ซึ่งถูกฟักบนตะแกรงเป็นเวลา 45 วินาที หลังจากขั้นตอนการซับสุดท้าย ตัวอย่างถูกปล่อยให้แห้งเป็นเวลาอย่างน้อย 20 นาทีก่อนที่จะดำเนินการถ่ายภาพด้วยกล้องจุลทรรศน์ FEI Tecnai 12 Bio-Twin ที่แรงดันไฟฟ้าความเร่งที่ 120 โวลต์
ไครโอ-อีเอ็ม
ตัวอย่างสำหรับไครโอ-EM ถูกเตรียมโดยใช้เครื่องทำให้แข็งตัว (Vitrobot, Thermo Fisher Scientific) ความเข้มข้นของ origami ในตัวอย่างที่ซับซ้อนคือ 90 nM สำหรับ 6HB-2k, 84 nM สำหรับ 24HB-2.5k, 18 nM สำหรับ 6HB-10k และ 21 nM สำหรับ 24HB-10k ส่งผลให้ความเข้มข้น CP ทั้งหมดเป็น 180 μM และ 210 μM สำหรับเชิงซ้อน ตัวอย่าง 6HB และ 24HB ตามลำดับ ส่วนที่เหลือ 3 ไมโครลิตรของตัวอย่างโอริกามิที่ซับซ้อนถูกฝากไว้บนตะแกรงที่เคลือบด้วยคาร์บอนโฮลีย์ (ทองแดง 50 mesh R002/200, Quantifoil) ที่ทำความสะอาดด้วยพลาสมา (1.2 วินาที, เครื่องมือ Harrick Plasma PDC-1.3-EC) หลังจากการฟักตัวเป็นเวลา 1 นาที ของเหลวส่วนเกินจะถูกซับเป็นเวลา 10 วินาทีที่ความชื้นสัมพัทธ์ 100% และ 6 °C ตามด้วยการจุ่มกริดลงในอีเทนเหลว กริดถูกเก็บไว้ในไนโตรเจนเหลว ข้อมูลถูกเก็บรวบรวมที่อุณหภูมิไนโตรเจนเหลวในกล้องจุลทรรศน์อิเล็กตรอนแบบส่องผ่าน Talos Arctica (Thermo Fisher Scientific) ทำงานที่ 200 kV โดยใช้เครื่องตรวจจับอิเล็กตรอนโดยตรง Falcon III (Thermo Fisher Scientific) ใช้กำลังขยาย 150,000× ส่งผลให้ขนาดพิกเซลที่ปรับเทียบแล้วอยู่ที่ 0.96 Å พารามิเตอร์การรวบรวมข้อมูลแสดงอยู่ในตารางเสริม 4 (หมายเหตุประกอบ 24).
การสร้างใหม่ด้วยอนุภาคเดี่ยว
ข้อมูล Cryo-EM ได้รับการประมวลผลโดยใช้ CryoSPARC 3.3.2 (เทคโนโลยีชีวภาพโครงสร้าง) เว้นแต่จะระบุไว้เป็นอย่างอื่น พารามิเตอร์ฟังก์ชันการถ่ายโอนคอนทราสต์ถูกประมาณค่าโดยใช้ CTFFIND4 (อ้างอิง 66). ส่วนตามเส้นใยถูกกำหนดโดยใช้ฟังก์ชัน Filament Tracer พารามิเตอร์สมมาตรของเฮลิคอลถูกประเมินเริ่มแรกจากค่าเฉลี่ยคลาส 2D โดยใช้ Helix Indexer ที่ใช้ Python67. โครงสร้างและพารามิเตอร์สมมาตรของลานได้รับการปรับปรุงโดยใช้ฟังก์ชัน Helix Refine และการปรับแต่งที่ไม่สม่ำเสมอในส่วนเกลียวที่แก้ไขการเคลื่อนไหว ในการกำหนดพารามิเตอร์สมมาตรแบบเฮลิคอลของชั้นนอก 6HB-10k การดำเนินการจำแนกประเภท 2D ครั้งที่สองได้ดำเนินการหลังจากลบการมีส่วนร่วมของชั้นในโดยใช้ฟังก์ชันการลบอนุภาค Helix Refine ทำงานบนเซตย่อยของอนุภาคที่แสดงชั้นที่สองที่ชัดเจน โดยใช้พารามิเตอร์สมมาตรที่กำหนดเป็นการประมาณเบื้องต้น การสร้างใหม่ถูกทำให้คมขึ้นโดยการใช้เฉพาะกิจ B-ปัจจัยของ −300 Å2. การสร้างใหม่ได้รับการเฉลี่ยในพื้นที่จริงโดยการกำหนดพารามิเตอร์สมมาตรของลานบนส่วนกลาง ซึ่งเป็นส่วนที่เรียงลำดับมากที่สุดของแผนที่ (50% ของปริมาตร) ใน Bsoft68.
สำหรับการสร้างแบบจำลองโครงสร้างของแคปโซเมอร์ CP โมโนเมอร์ (PDB:1cwp) ได้รับการติดตั้งในการสร้างใหม่ 6HB-2k ในตำแหน่งหกตำแหน่งของเฮกซาเมอร์ในฐานะตัวแข็งใน UCSF ChimeraX 1.3 (อ้างอิง 69). แบบจำลองอะตอมได้รับการปรับปรุงเทียบกับความหนาแน่นโดยใช้ ISOLDE 1.3 (อ้างอิง 70) และ Phenix 1.19 (อ้างอิง 71). ในการสร้างการแสดงอะตอมของเส้นใย จึงมีการสร้างสำเนาสมมาตรของเฮกซาเมอร์ใน ChimeraX เพื่อให้เห็นภาพตำแหน่งของเฮกซาเมอร์และเพนทาเมอร์ของ CP ในฝาครอบ ได้มีการเลือกฝาครอบของเส้นใย 6HB-2k ด้วยตนเองในไมโครกราฟ โครงสร้างฝาครอบได้รับการปรับปรุงโดยใช้ฟังก์ชัน Helix Refine โดยละเว้นความสมมาตร เนื่องจากทำให้สามารถจำกัดมุมเอียงของฝาครอบใกล้กับมุมมองด้านข้างได้ การสร้างฝาครอบขึ้นมาใหม่ถูกกรองตามความละเอียดเฉพาะที่โดยใช้ตัวกรองเฉพาะที่ แบบจำลองอะตอมเฮกซาเมอร์และโครงสร้างเพนทาเมอร์ที่กำหนดไว้ก่อนหน้านี้ (สกัดจาก PDB:1cwp หลังจากใช้สมมาตรไอโคซาฮีดรัล) ได้รับการติดตั้งเป็นวัตถุแข็งใน ChimeraX 1.3 พารามิเตอร์การประมวลผลข้อมูลได้รับในตารางเสริม 4. พารามิเตอร์การปรับแต่งและการตรวจสอบแบบจำลองจะแสดงในตารางเสริม 5.
แซ็กซ์
ตัวอย่างสำหรับ SAXS ถูกจัดทำขึ้นที่ความเข้มข้นของ origami ที่ 165 nM (6HB ซึ่งสอดคล้องกับความเข้มข้น CP ที่แยกชิ้นส่วนที่ 330 μM) และ 180 nM (24HB ซึ่งสอดคล้องกับความเข้มข้นของ CP ที่แยกชิ้นส่วนที่ 450 μM) และปิดผนึกภายในเส้นผ่านศูนย์กลาง 1.5 มม. เส้นเลือดฝอยแก้ว การวัดดำเนินการโดยใช้อุปกรณ์ Xenocs Xeuss 3.0C ที่มาพร้อมกับแหล่งกำเนิดไมโครโฟกัสทองแดง GeniX 3D (ความยาวคลื่น λ = 1.542 Å) และตัวตรวจจับพิกเซลแบบไฮบริด EIGER2 R 1M ที่ระยะห่างระหว่างตัวตรวจจับตัวอย่าง 1,100 มม. ดำเนินการเก็บข้อมูลเป็นเวลา 3 × 3 ชั่วโมงต่อตัวอย่าง ในการรับข้อมูล 1D SAXS ข้อมูลการกระเจิง 2D จะถูกหาค่าเฉลี่ยแบบอะซิมัททัล ขนาดของเวกเตอร์การกระเจิง q ได้รับจาก (q,=,4uppi sin theta /แลมบ์ดา) กับ2θ เป็นมุมกระเจิง การบำบัดข้อมูลรวมถึงการหาค่าเฉลี่ยของข้อมูล 2D เพิ่มขึ้นสามเท่าของแต่ละตัวอย่าง การลบพื้นหลังออกจากบัฟเฟอร์เชิงซ้อน (3.25 มิลลิโมลาร์ HEPES-NaOH, 25 มิลลิโมลาร์ Tris–HCl, 150 มิลลิโมลาร์ NaCl, 0.5 มิลลิโมลาร์ DTT) และฟอร์มแฟคเตอร์ถูกติดตั้งเข้ากับกระบอกสูบ ( 6HB, 24HB), ทรงกลม (T = ชุดประกอบ icosahedral CPs 3 ชุด) และกระบอกสูบ core–shell (6HB-2k, 24HB-2.5k) โดยใช้ซอฟต์แวร์ SasView มีการเพิ่มโมเดล Debye–Anderson–Brumberger เพื่อรองรับพื้นหลัง
- เนื้อหาที่ขับเคลื่อนด้วย SEO และการเผยแพร่ประชาสัมพันธ์ รับการขยายวันนี้
- PlatoData.Network Vertical Generative Ai เพิ่มพลังให้กับตัวเอง เข้าถึงได้ที่นี่.
- เพลโตไอสตรีม. Web3 อัจฉริยะ ขยายความรู้ เข้าถึงได้ที่นี่.
- เพลโตESG. ยานยนต์ / EVs, คาร์บอน, คลีนเทค, พลังงาน, สิ่งแวดล้อม แสงอาทิตย์, การจัดการของเสีย. เข้าถึงได้ที่นี่.
- BlockOffsets การปรับปรุงการเป็นเจ้าของออฟเซ็ตด้านสิ่งแวดล้อมให้ทันสมัย เข้าถึงได้ที่นี่.
- ที่มา: https://www.nature.com/articles/s41565-023-01443-x
- :มี
- :เป็น
- ][หน้า
- 1
- 1 อัตราส่วน 1
- 1.3
- 10
- 100
- 11
- 116
- 12
- 14
- 15%
- 16
- 180
- 19
- 1M
- 2%
- 20
- 200
- 2011
- 2014
- 2015
- 2018
- 2019
- 2021
- 2022
- 2023
- 23
- 24
- 25
- 27
- 2D
- 30
- 31
- 32
- 3d
- 40
- 46
- 50
- 500
- 60
- 65
- 66
- 67
- 7
- 70
- 75
- 8
- 80
- 84
- 9
- 90
- a
- ข้างบน
- การเร่งความเร็ว
- ลงชื่อเข้าใช้
- ถูกต้อง
- ที่ได้มา
- การครอบครอง
- Ad
- เหมาะ
- ที่เพิ่ม
- นอกจากนี้
- เพิ่มเติม
- นอกจากนี้
- ปรับ
- การปรับ
- หลังจาก
- กับ
- อายุ
- ตัวแทน
- AIR
- AL
- อนุญาตให้
- ตาม
- an
- การวิเคราะห์
- สมอ
- และ
- การประยุกต์ใช้
- ใกล้เข้ามา
- เป็น
- รอบ
- AS
- ลอม
- การชุมนุม
- At
- ค่าเฉลี่ย
- ไป
- พื้นหลัง
- ตาม
- BE
- รับ
- ก่อน
- กำลัง
- ระหว่าง
- ผูกพัน
- เทคโนโลยีชีวภาพ
- ร่างกาย
- ทั้งสอง
- BP
- สั้น
- กันชน
- การก่อสร้าง
- by
- CAN
- ฝาครอบ
- หมวก
- เซลล์
- โทรศัพท์มือถือ
- ส่วนกลาง
- ช่อง
- ทางเลือก
- ชั้น
- การจัดหมวดหมู่
- ชัดเจน
- คลิก
- ปิดหน้านี้
- ชุด
- การแข่งขัน
- สมาธิ
- เงื่อนไข
- มี
- อย่างต่อเนื่อง
- ตรงกันข้าม
- ผลงาน
- การควบคุม
- สำเนา
- ทองแดง
- ตรงกัน
- สร้าง
- ที่สร้างขึ้น
- ข้อมูล
- การประมวลผล
- กำหนด
- การจัดส่ง
- ทั้งนี้ขึ้นอยู่กับ
- ฝาก
- อธิบาย
- ได้รับการออกแบบ
- ที่ต้องการ
- กำหนด
- แน่นอน
- นักพัฒนา
- เครื่อง
- การล้างไต
- ต่าง
- Dimension
- โดยตรง
- ระยะทาง
- ดีเอ็นเอ
- ลง
- แห้ง
- ในระหว่าง
- พลวัต
- e
- E&T
- แต่ละ
- ed
- นักการศึกษา
- ผล
- ปลาย
- ชั้นเยี่ยม
- ทำให้มั่นใจ
- สิ่งแวดล้อม
- เท่ากัน
- พร้อม
- ประมาณ
- ประมาณการ
- อีเธอร์ (ETH)
- ตัวอย่าง
- ส่วนเกิน
- ตลาดแลกเปลี่ยน
- แลกเปลี่ยน
- การแสดงออก
- การสูญพันธุ์
- ปัจจัย
- เหยี่ยวนกเขา
- FAST
- fei
- กรอง
- ฟิลเตอร์
- สุดท้าย
- หา
- ชื่อจริง
- แฟลช
- ตาม
- สำหรับ
- ฟอร์ม
- การสร้าง
- สี่
- ราคาเริ่มต้นที่
- ฟังก์ชัน
- นอกจากนี้
- กำหนด
- กระจก
- ค่อยๆ
- ตะแกรง
- การเจริญเติบโต
- แนวทาง
- มี
- โปรดคลิกที่นี่เพื่ออ่านรายละเอียดเพิ่มเติม
- จุดสูง
- อย่างไรก็ตาม
- HTTPS
- เป็นลูกผสม
- i
- ICE
- ICON
- iii
- ภาพ
- ภาพ
- การถ่ายภาพ
- ทันที
- ท่วม
- การแช่
- การจัดเก็บภาษี
- in
- รวม
- เพิ่มขึ้น
- ฟักไข่
- ฟักไข่
- ตัวทำดัชนี
- แรกเริ่ม
- ในขั้นต้น
- ตราสาร
- แบบบูรณาการ
- ปฏิสัมพันธ์
- อินเตอร์เฟซ
- เข้าไป
- อิออน
- เปลี่ยว
- IT
- ITS
- KDA
- ชั้น
- น้อยที่สุด
- ซ้าย
- เบา
- LINK
- ของเหลว
- จดทะเบียน
- โหลด
- ในประเทศ
- สำคัญ
- ด้วยมือ
- แผนที่
- แผนที่
- วัสดุ
- วัด
- วัด
- การวัด
- ตาข่าย
- วิธีการ
- ไมกา
- กล้องจุลทรรศน์
- กล้องจุลทรรศน์
- นาที
- ผสม
- สารผสม
- การเคลื่อนย้าย
- โหมด
- แบบ
- การสร้างแบบจำลอง
- โมดูลาร์
- MOL
- โมเลกุล
- การตรวจสอบ
- มากที่สุด
- mRNA
- นาโน
- นาโนเทคโนโลยี
- ธรรมชาติ
- เชิงลบ
- ได้รับ
- of
- on
- การดำเนินการ
- การปรับให้เหมาะสม
- มิฉะนั้น
- ผล
- ค้างคืน
- ออกซิเจน
- กระดาษ
- พารามิเตอร์
- ส่วนหนึ่ง
- อนุภาค
- พีบีเอส
- PCR
- ต่อ
- ดำเนินการ
- ทางร่างกาย
- เลือก
- ขว้าง
- พิกเซล
- การวาง
- พลาสมา
- เวที
- เพลโต
- เพลโตดาต้าอินเทลลิเจนซ์
- เพลโตดาต้า
- ซึ่งพรวดพราด
- ตำแหน่ง
- การจัดเตรียม
- เตรียม
- นำเสนอ
- ก่อนหน้านี้
- เชื้อปะทุ
- PROC
- ขั้นตอน
- การประมวลผล
- การประมวลผล
- โครงการ
- การป้องกัน
- โปรตีน
- โปรตีน
- ซื้อ
- คุณภาพ
- ตั้งแต่
- อัตราส่วน
- มาถึง
- ถึง
- ปฏิกิริยา
- จริง
- เหมือนจริง
- สีแดง
- ปรับแต่ง
- กลั่น
- เกี่ยวกับ
- ญาติ
- ซ้ำแล้วซ้ำอีก
- รายงาน
- จำเป็นต้องใช้
- นักวิจัย
- ความละเอียด
- ตามลำดับ
- ส่งผลให้
- ผลสอบ
- เข้มงวด
- อาร์เอ็นเอ
- ห้อง
- วิ่ง
- s
- เกลือ
- SCI
- วิทยาศาสตร์
- วิทยาศาสตร์
- ที่สอง
- เห็น
- กลุ่ม
- เปลี่ยน
- สั้น
- แสดงให้เห็นว่า
- แสดง
- ด้าน
- เหมือนกับ
- เว็บไซต์
- หก
- ขนาด
- มีขนาดเล็กกว่า
- โซเดียม
- ซอฟต์แวร์
- ทางออก
- โซลูชัน
- แหล่ง
- ช่องว่าง
- แยก
- ระบุ
- คงที่
- ขั้นตอน
- ขั้นตอน
- สต็อก
- การเก็บรักษา
- เก็บไว้
- เส้น
- กระแส
- ความแข็งแรง
- โครงสร้าง
- การศึกษา
- ศึกษา
- ต่อจากนั้น
- ที่ประสบความสำเร็จ
- ระบบ
- ตาราง
- ลอส
- เทคโนโลยี
- ที่
- พื้นที่
- ของพวกเขา
- แล้วก็
- theta
- นี้
- สาม
- ตลอด
- เวลา
- ครั้ง
- ไปยัง
- ร่วมกัน
- เครื่องมือ
- รวม
- ผู้ตามรอย
- โอน
- โอน
- การรักษา
- กลับ
- สอง
- ภายใต้
- สหรัฐอเมริกา
- ใช้
- มือสอง
- ผู้ใช้งาน
- ส่วนติดต่อผู้ใช้
- การใช้
- วัคซีน
- ตรวจสอบความถูกต้อง
- การตรวจสอบ
- อเนกประสงค์
- ยอดวิว
- ไวรัส
- การสร้างภาพ
- แรงดันไฟฟ้า
- ปริมาณ
- ไดรฟ์
- W
- คือ
- การซัก
- น้ำดื่ม
- น้ำหนัก
- คือ
- ที่
- ขาว
- กับ
- ภายใน
- ไม่มี
- X
- อัตราผลตอบแทน
- ลมทะเล