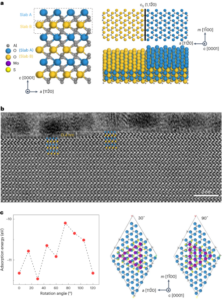
Synteza nanobotów
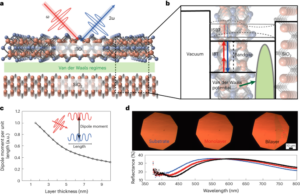
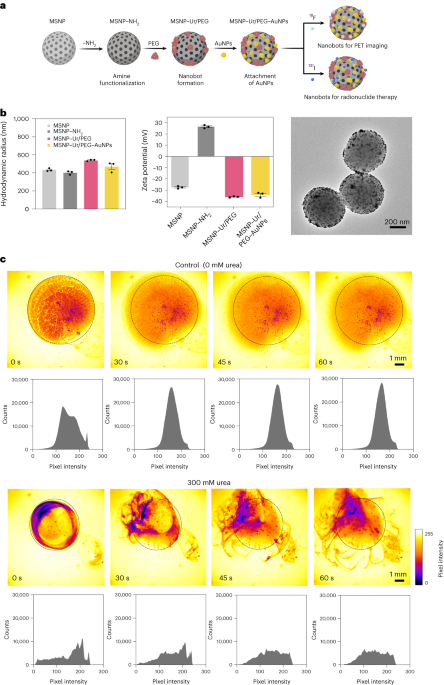
Nanoboty przygotowano w sposób opisany wcześniej33. W skrócie, MSNP zsyntetyzowano przy użyciu zmodyfikowanej metody Stöbera41poddając reakcji trietanoloaminę (35 g), ultraczystą wodę (20 ml) i bromek heksadecylotrimetyloamoniowy (CTAB; 570 mg) w temperaturze 95 °C przez 30 min, mieszając. Następnie wkroplono ortokrzemian tetraetylu (1.5 ml); mieszaninę pozostawiono do przereagowania na 2 godziny w temperaturze 95 °C, a powstałe MSNP zebrano przez odwirowanie i przemyto etanolem (trzy razy, 2,500g, 5 minut). Aby usunąć matrycę CTAB, MSNP umieszczono pod chłodnicą zwrotną w kwaśnym metanolu (1.8 ml HCl, 30 ml metanolu) na 24 h. Następnie MSNP zebrano przez odwirowanie i trzykrotnie przemyto etanolem (2,500 µl).g, 5 min) przed wprowadzeniem modyfikacji aminowej poprzez dodanie APTES (6 µl na mg MSNP) do MSNP (1 mg ml-1) w 70% roztworze etanolu w temperaturze 70 °C, energicznie mieszając przez 1 h. MSNP-NH2 zebrano i przemyto trzykrotnie etanolem i trzykrotnie wodą przez odwirowanie (trzykrotnie po 1,150g, 5 minut). MSNP-NH2 zawieszono ponownie w PBS w stężeniu 1 mg ml-1 i całkowitej objętości 900 µl i aktywowano aldehydem glutarowym (100 µl) przez 2.5 h w temperaturze pokojowej. Aktywowane MSNP-NH2 zebrano i przemyto trzykrotnie w PBS przez odwirowanie (1,150 ?g, 5 min), ponownie zawiesza się w roztworze ureazy (3 mg ml-1) i heterobifunkcjonalny PEG (1 μg PEG na mg 5 kDa HS-MSNPs-NH2) w PBS i poddano reakcji przez 24 godziny w temperaturze pokojowej. Powstałe nanoboty następnie zebrano i przemyto trzykrotnie w PBS przez wirowanie (1,150 ?g, 5 min) przed ponownym zawieszeniem ich w dyspersji AuNP, przygotowanej jak opisano wcześniej51, pozostawiając je na 10 minut i dokładnie przemywając przez odwirowanie (trzy razy, 1,150g, 5 minut).
Hydrodynamiczny rozkład wielkości i ładunek powierzchniowy MSNP, MSNP-NH2, nanoboty i nanoboty ozdobione AuNP określono odpowiednio przy użyciu systemu dynamicznego rozpraszania światła Wyatt Mobius i Malvern Zetasizer. We wszystkich przypadkach stężenie wynosiło 20 µg ml-1 i czas akwizycji 5 s, przy użyciu trzech serii na eksperyment. Przeprowadzono trzy pomiary dla każdego rodzaju cząstek.
Synteza MSNP FITC
Przygotowano mieszaninę FITC (2 mg), etanolu (5 ml) i APTES (400 µl) i mieszano przez 30 min. Następnie postępowano zgodnie z wcześniej opisanym protokołem syntezy MSNP, z tą różnicą, że dodaliśmy kroplami ortokrzemian tetraetylu (1.25 ml) w połączeniu z mieszaniną FITC – APTES (250 µl). Etapy funkcjonalizacji prowadzące do otrzymania nanobotów znakowanych FITC były takie, jak opisano powyżej.
Synteza AuNP
AuNP zsyntetyzowano przy użyciu opisanej metody33. Krótko mówiąc, wszystkie materiały zostały oczyszczone przy użyciu świeżo przygotowanej wody królewskiej, dokładnie spłukane wodą i wysuszone na powietrzu. Następnie 1 mM AuCl4 roztwór ogrzano do temperatury wrzenia, mieszając, w kolbie okrągłodennej zintegrowanej z systemem refluksu. Następnie dodano 10 ml roztworu cytrynianu sodu (30.8 mM) i roztwór gotowano przez 20 min, uzyskując czerwony kolor. Następnie roztwór pozostawiono do ochłodzenia do temperatury pokojowej, mieszając przez 1 godzinę. Powstałe AuNP przechowywano w ciemności i przeprowadzono charakterystykę za pomocą transmisyjnej mikroskopii elektronowej.
Aktywność enzymatyczna
Aktywność enzymatyczna nanobotów, 18F-nanoboty i 131Pomiary I-nanobotów przeprowadzono przy użyciu czerwieni fenolowej. Aby to zrobić, 2 µl nanobotów (1 mg ml-1) dodano do 96-studzienkowej płytki i zmieszano z 200 µl różnych roztworów mocznika (0, 50, 100, 200 mM) w 1.1 mM czerwieni fenolowej. Absorbancję przy 560 nm mierzono w czasie w temperaturze 37 °C.
Dynamika ruchu nanobotów w mikroskopii optycznej
Optyczne filmy przedstawiające nanoboty zarejestrowano przy użyciu mikroskopu Leica Thunder w połączeniu z szybką kamerą CCD Hamamatsu i obiektywem × 1.25. W tym celu nanoboty odwirowano i ponownie zawieszono w 50 µl PBS (końcowe stężenie 20 mg ml-1). Następnie szalkę Petriego napełniono 3 ml PBS lub 300 mM roztworem mocznika (w PBS) i obserwowano pod mikroskopem. Kropla 5 µl z nanobotami (20 mg ml-1) następnie dodano do wypełnionej płynem szalki Petriego i nagrywano filmy z szybkością 25 klatek na sekundę. Rozkłady intensywności pikseli wideo w obszarach ROI analizowano w odstępach 15-sekundowych przy użyciu oprogramowania ImageJ.
Radioznakowanie nanobotów za pomocą [18F]F-PyTFP
Synteza [18F]F-PyTFP
[18F]F-PyTFP zsyntetyzowano w module Neptis xSeed (Optimized Radiochemical Applications), zgodnie z wcześniej opisaną metodą33.
Synteza 18Nanoboty oznaczone literą F
Nanoboty oznaczono [18F]F-PyTFP, w oparciu o ustaloną wcześniej procedurę z niewielkimi modyfikacjami33. W skrócie, 200 µl roztworu nanobota (1 mg ml-1) odwirowano (10 min, 13,853g), ponownie zawieszono w 10 µl PBS (1 mM, pH 8) i inkubowano z 4 µl [18F]F-PyTFP w acetonitrylu (około 37 MBq) przez 35 min w temperaturze pokojowej. Po inkubacji mieszaninę reakcyjną rozcieńczono wodą (200 µl) i oczyszczono przez wirowanie (5 min, 13,853g). Powstały osad następnie przepłukano trzykrotnie wodą przed zmierzeniem w kalibratorze dawki (CPCRC-25R, Capintec). Wydajność radiochemiczną obliczono jako stosunek ilości radioaktywności obecnej w nanobotach po umyciu do początkowej ilości radioaktywności. Czystość radiochemiczna po oczyszczeniu wyniosła ≥99%, jak określono za pomocą radiowej chromatografii cienkowarstwowej (radio-TLC) przy użyciu bibuły chromatograficznej iTLC-SG (Agilent Technologies) oraz dichlorometanu i metanolu (2:1) jako odpowiednio fazy stacjonarnej i ruchomej. Płytki TLC analizowano przy użyciu czytnika TLC (MiniGITA, Raytest).
Stabilność 18F-nanoboty
Stabilność 18Nanoboty znakowane F oznaczono przy użyciu następujących pożywek: (1) 300 mM mocznika, (2) wody i (3) moczu zwierząt z nowotworem. 18Nanoboty znakowane F (10 µl) inkubowano z odpowiednim roztworem (100 µl) przez 1 h w temperaturze pokojowej. Następnie nanoboty i supernatant oddzielono przez odwirowanie i zebrano, a radioaktywność zmierzono w kalibratorze dawki (CPCRC-25R).
Radioznakowanie nanobotów za pomocą 131I
Radiojodowanie nanobotów ureazowych przeprowadzono poprzez inkubację nanobotów z wstrzykiwalnym [131Roztwór I]NaI (925 MBq ml-1; GE HealthCare). W skrócie, 400 µl roztworu nanobota ureazy (1 mg ml-1) odwirowano (13,853g, 5 min), ponownie zawieszono w 100 µl PBS (10 mM, pH 7.4) i inkubowano z 25 µl lub 185 µl wstrzykiwalnego [131I]NaI (odpowiednio około 42.55 lub 277.5 MBq) przez 30 min, w zależności od pożądanej aktywności końcowej. Po inkubacji mieszaninę reakcyjną oczyszczono przez wirowanie (13,853g, 5 minut). Powstały osad przemyto trzykrotnie wodą (100 µl). Radioaktywność w supernatancie i osadzie oznaczono za pomocą kalibratora dawki (CPCRC-25R), a obie frakcje analizowano metodą radio-TLC, podobnie jak w przypadku 18F-nanoboty.
Rozwój modelu zwierzęcego
Myszy utrzymywano i traktowano zgodnie z dyrektywą Rady Europejskiej 2010/63/UE i wewnętrznymi wytycznymi. Wszystkie procedury eksperymentalne zostały zatwierdzone przez komisję etyczną CIC biomaGUNE i władze lokalne (Diputación Foral de Guipuzcoa, PRO-AE-SS-276). Analiza obrazu (zarówno PET, jak i MRI) nie uwzględniała rozmieszczenia zwierząt w grupach.
Ortotopowy mysi model raka pęcherza wygenerowano przez dopęcherzowe podanie komórek MB49 (linia komórkowa mysiego raka pęcherza moczowego) samicom myszy C57BL/6JRj (8 tygodni, Janvier). Do eksperymentów mających na celu określenie akumulacji nowotworu (cztery grupy; szczegóły poniżej) zaszczepiono sześć zwierząt na grupę, jak określono za pomocą analizy dokładności, przy następujących założeniach: wymagana precyzja, 20%; oczekiwane odchylenie standardowe, ±20%; pewność siebie, 95%; strata zwierząt, 20%. Do eksperymentów dotyczących skuteczności terapeutycznej (sześć grup; szczegóły poniżej) włączono po dziesięć zwierząt na grupę, jak obliczono za pomocą jednostronnego testu Studenta t-test, różnica pomiędzy dwoma niezależnymi średnimi, przy następujących założeniach: hipoteza zerowa, leczenie nie wpływa na wzrost guza; α, 0.05; 1 − β, 0.95; sd, ±50%; oczekiwane różnice między grupami, 50%; strata zwierząt, 20%. Ponieważ ze względów operacyjnych doświadczenie przeprowadzono w dwóch seriach, do obu serii włączono po jednej grupie kontrolnej (tab 2), a następnie wszystkie zwierzęta połączono. W celu ustalenia guza myszy znieczulono poprzez wdychanie 3% izofluranu w czystym O2 i utrzymywany 1.0–1.5% izofluranem w 100% O2. Następnie opróżniono pęcherz i wywołano zmiany chemiczne na nabłonku dróg moczowych poprzez dopęcherzowe wkroplenie 50 µl poli-l-lizyna (Sigma-Aldrich) przez cewnik o średnicy 24 G przez 15 minut. Następnie ponownie opróżniono pęcherz i pobrano komórki MB49 (105 komórek) w DMEM o wysokiej zawartości glukozy (100 µl) wkroplono na 1 h przed wyjęciem cewnika i opróżnieniem pęcherza poprzez masaż brzucha. Przez cały czas trwania eksperymentów myszy były monitorowane i ważone w celu monitorowania stanu zdrowia i dobrostanu. Ludzki punkt końcowy zastosowano, jeśli utrata masy ciała przekroczyła 20% lub na podstawie objawów klinicznych, zgodnie z kryteriami lekarza weterynarii.
Śledzenie wielkości guza
Badania MRI przeprowadzono 7 i 14 dni po indukcji nowotworu, przy użyciu skanera 7 T Bruker BioSpec USR 70/30 (Bruker BioSpin) wyposażonego w gradientową wstawkę BGA-12S o wartości 440 mT m-1 oraz rezonator 112/086 QSN (T12053V3) dla częstotliwości radiowej14 transmisji oraz cewkę powierzchniową mózgu szczura (T11205V3) do odbioru częstotliwości radiowej (obie pracujące na częstotliwości 300 MHz). Zwierzęta znieczulono izofluranem (4% do indukcji i 1.5% do podtrzymania w 50% OXNUMX).2/50% N2 mieszaniny) i umieścić na podstawce kompatybilnej z MR. Temperaturę ciała i częstość oddechów monitorowano w sposób ciągły za pomocą urządzenia monitorującego kompatybilnego z MR (model 1030 SA, Small Animal Instruments), połączonego z systemem podgrzewacza powietrza dla małych gryzoni w celu utrzymania temperatury ciała. Po uzyskaniu obrazów referencyjnych do obrazowania nowotworów zastosowano sekwencję obrazowania ważoną dyfuzyjnie opartą na echu spinowym, stosując następujące parametry: czas echa (TE) = 22.3 ms, czas powtarzania (TR) = 2,500 ms, n = 2 średnie, jeden obraz A0 (obraz podstawowy z b = 0 s mm-2) i jeden obraz DW uzyskany przy użyciu gradientów dyfuzji w kierunku (1, 0, 0) z czasem trwania gradientu δ = 4.5 ms i separacja gradientowa Δ = 10.6 ms, daje b = 650 s mm-2, 16 × 16 mm2 pole widzenia, rozmiar matrycy obrazu 160 × 160 punktów, 20 kolejnych warstw o grubości 0.5 mm (bez przerwy, uzyskane w trybie przeplatanym) i szerokość pasma 192.9 Hz na piksel. Aby zwizualizować guzy, obrazy poddano obróbce końcowej za pomocą oprogramowania ImageJ, dzieląc obrazy uzyskane za pomocą gradientu dyfuzji (b = 650 s mm-2) przez te nabyte bez (b = 0 s mm-2) i zastosowanie trójwymiarowego filtra Gaussa (σx = σy = σz = 0.7) do wyniku. Guzy zaznaczono ręcznie w celu określenia ich objętości.
Biodystrybucja in vivo
W 15 dniu po indukcji nowotworu myszy podzielono losowo na cztery grupy w celu uzyskania jednorodnego średniego rozkładu objętości guza pomiędzy grupami. Skany PET-CT (skanery MOLECUBES β i X-CUBE) wykonano 3 h po dopęcherzowym podaniu 100 µl 18F-BSA (grupy 1 i 2) lub 18Nanoboty F-ureazy (grupy 3 i 4) w stężeniu 200 µg ml-1, stosując jako nośnik wodę (grupy 1 i 3) lub 300 mM mocznika w wodzie (grupy 2 i 4) (tabela 1). W celu uzyskania obrazu zwierzęta wprowadzono do znieczulenia (5% izofluran w czystym tlenie) i ułożono w pozycji leżącej przed masowaniem okolicy brzucha w celu opróżnienia pęcherza. Zaraz potem odpowiedni 18Nanoboty oznaczone literą F (18F-BSA/18F-ureaza w wodzie/moczniku) wkroplono do pęcherza przez cewnik o rozmiarze 24 G i inkubowano przez 1 godzinę, po czym usunięto cewnik, opróżniono pęcherz i pozostawiono myszy do wyzdrowienia po znieczuleniu. Na t = 3 h po podaniu zwierzęta ponownie znieczulono i po 10 min uzyskano statyczne obrazy PET całego ciała, a następnie wykonano tomografię komputerową. Obrazy PET zrekonstruowano przy użyciu algorytmu rekonstrukcji maksymalizacji oczekiwań podzbioru uporządkowanego 3D z korektami losowymi, rozproszenia i tłumienia. Obrazy PET-CT tej samej myszy zostały wspólnie zarejestrowane i przeanalizowane przy użyciu narzędzia do przetwarzania obrazu PMOD. Wykresy stężenia radioaktywności w funkcji czasu uzyskano poprzez utworzenie interesującej objętości w górnym obszarze pęcherza przy użyciu narzędzia konturowego 3D i zmierzenie aktywności (skorygowanej za rozpad) w kilobekerelach na narząd. Wyniki skorygowano poprzez zastosowanie współczynnika kalibracyjnego, a następnie znormalizowano w oparciu o objętość guza uzyskaną z rezonansu magnetycznego.
Badania ex vivo
Analizy histopatologiczne
Po wykonaniu wszystkich badań obrazowych wybrane pęcherze (n = 3 na grupę) od zwierząt z nowotworem i zdrowych usunięto w warunkach aseptycznych i natychmiast utrwalono w 4% formaldehydzie. Następnie pęcherze zatopiono w parafinie przed pobraniem skrawków o wielkości 2–3 µm do barwienia hematoksyliną i eozyną. Do badania histopatologicznego uzyskano reprezentatywne obrazy we wszystkich warunkach.
Analiza ICP-MS
Pomiary przeprowadzono na urządzeniu Thermo iCAP Q ICP-MS (Thermo Fisher Scientific) w połączeniu z autosamplerem ASX-560 (CETAC Tech). Po wykonaniu wszystkich badań obrazowych zwierzęta uśmiercono, a wybrane pęcherze (n = 2 na grupę; cztery grupy) zebrano i strawiono w 1 ml HNO3:HCl (mieszanina 4:1). Dyspersję gotowano aż do całkowitego rozpuszczenia narządów. Następnie roztwór ochłodzono do temperatury pokojowej i poddano analizie za pomocą ICP-MS w celu określenia stężenia Au w każdej próbce, przeliczając wyniki na procent wstrzykniętej dawki na gram tkanki (%ID g-1).
Immunohistochemia i obrazowanie za pomocą mikroskopu konfokalnego
Do analiz immunohistochemicznych zwierzęta z nowotworem otrzymywały nanoboty znakowane FITC w wodzie lub 300 mM moczniku (n = 4 na grupę), jak opisano powyżej, dla badań PET-CT. Dodatkowo zwierzęta z nowotworem bez nanobotów służyły jako grupa kontrolna (n = 2). We wszystkich przypadkach pęcherze zebrano, zamrożono i pocięto na skrawki o średnicy 10 µm, które natychmiast utrwalono w 10% formaldehydzie przez 15 min, przemyto 10 mM PBS, a następnie inkubowano w 50 mM NH4Cl w PBS przez 5 minut, po czym ponownie przemyto PBS. Permeabilizację przeprowadzono za pomocą metanolu:acetonu (1:1) przez 5 minut w temperaturze pokojowej i 0.1% Triton w PBS przez 5 minut. Po przemyciu PBS próbki nasycano roztworem 5% BSA – 0.5% Tween w PBS przez 15 minut w temperaturze pokojowej i inkubowano przez 1 godzinę w temperaturze pokojowej z mysim anty-FITC (1:100, Abcam) w 5% BSA –0.5% Tween. Skrawki przemyto trzykrotnie 10 mM PBS przez 5 min i inkubowano przez 30 min w temperaturze pokojowej z przeciwciałem drugorzędowym Alex Fluor 647 osła przeciwko mysim IgG (Molecular Probes, Life Technologies, 1:1,000) w 5% BSA–0.5% Tween w PBS, przemyto ponownie w PBS (3 × 5 min) i zamontowano za pomocą zestawu zapobiegającego blaknięciu ProLong z 4,6-diamidyno-2-fenyloindolem (DAPI; Molecular Probes, Life Technologies). Obrazy uzyskano za pomocą mikroskopu konfokalnego Leica STELLARIS 5 (Park Naukowy UPV/EHU) przy identycznych ustawieniach dla wszystkich przekrojów: powiększenie × 10 z obrazowaniem płytek i szyciem (zwykle pole widzenia 4 × 5). Linia lasera i okna detekcji wynosiły 405 nm i 440–503 nm dla DAPI, 489 nm i 494–602 nm dla białego lasera FITC oraz 653 nm i 660–836 nm dla białego lasera Alexa647.
Oczyszczanie optyczne
Po perfuzji 4% paraformaldehydem i PBS próbki pęcherza moczowego usunięto i utrwalono w 4% paraformaldehydzie przez noc w temperaturze 4 °C, a następnie osadzono w 5 ml strzykawce z 0.8% agarozą o niskiej temperaturze topnienia w celu utworzenia cylindrycznego bloku umożliwiającego łatwe montaż w kuwecie kwarcowej. Cały blok stopniowo odwadniano stosując metanol:H2O w 4 °C (30%:70% przez 1 h, 50%:50% przez 1 h, 70%:30% przez 1 h, 100%:0% przez 1 h, następnie 100% metanol przez noc i ponownie przez noc 4 h) i na koniec zanurzono w alkoholu benzylowym i benzoesanie benzylu (BABB) jako roztworze dopasowującym współczynnik załamania światła do obrazowania. Do porównań in vitro zielonych nanobotów FITC z dostępnymi na rynku czerwonymi cząsteczkami użyliśmy czerwonych fluorescencyjnych nanocząstek krzemionki DiagNano (Creative Diagnostics) o średnicy 1 µm, odpornych na czyszczenie BABB.
Obrazowanie autofluorescencji i spolaryzowanego sLS
Obrazowanie arkusza świetlnego przeprowadzono na MacroSPIM, niestandardowym systemie do czystego obrazowania całych narządów opracowanym w IRB Barcelona44,45. W skrócie, próbki umieszcza się w bloku agarozowym, oczyszcza wraz z próbką i obrazuje w kuwecie kwarcowej. W obrazowaniu autofluorescencyjnym wykorzystano lasery o długości fali 488, 561 lub 638 nm zapewniające oświetlenie przez achromatyczną podwójną soczewkę cylindryczną o średnicy 50 mm (ACY254-050-A, Thorlabs). Aby zredukować artefakty w postaci pasków, arkusz świetlny obraca się za pomocą skanera rezonansowego SC-10 (EOPC) wzdłuż teleskopu 4f z achromatycznymi soczewkami dubletowymi G322288322 100 mm (QI Optic Photonics). Autofluorescencję tkanki zbiera się za pomocą pasmowych lub długoprzepustowych filtrów fluorescencyjnych i rejestruje za pomocą kamery ORCA Flash v2 (Hamamatsu Photonics). Obrazowanie przeprowadzono przy × 9.6, 8 z zoomem × 2, obiektywem × 0.6 i obiektywem tubusowym × 5, 6. Arkusz świetlny został spłaszczony w całym polu widzenia, uzyskując rozdzielczość osiową 3–2.5 µm. Obrazowanie 2D wykonywano w krokach co 3 µm. Obrazowanie całego pęcherza wykonano w 3 × 4 lub XNUMX × XNUMX XY płytki, w zależności od wielkości organów.
Obrazowanie sLS uzyskano poprzez usunięcie filtra fluorescencyjnego lub zastosowanie dowolnego filtra przepuszczającego laser. Obracanie arkusza świetlnego zmniejszyło szum plamek lasera, co skutkowało czasowym uśrednieniem spójności lasera, jak pokazano wcześniej52. Orientację liniowej polaryzacji arkusza świetlnego w oświetleniu kontrolowano poprzez obracanie płytki półfalowej (AHWP05M-600, Thorlabs) przed skanerem obrotowym. Wykryty sygnał selekcjonowano w polaryzacji przy użyciu obrotowego polaryzatora liniowego (LPVISC100, Thorlabs) przed kołem filtrów w detekcji, wprowadzając >50% utratę intensywności w detekcji fluorescencji. Podczas gdy rozkład sygnału sLS ogólnie zmienia się wraz z orientacją polaryzatora, obrót polaryzatora nie ma wpływu na sygnał autofluorescencji tkanki. sLS daje rozdzielczość przestrzenną 2.4 ± 0.3 µm w BABB, która jest porównywalna z rozdzielczością obrazowania fluorescencyjnego arkusza świetlnego (potwierdzona przez dopasowanie funkcji Gaussa do XY odpowiedź obrazu pojedynczej cząstki, rys. uzupełniająca. 8l–m) i zbliżoną do teoretycznej rozdzielczości w powietrzu (1.53 µm przy aperturze numerycznej (NA) = 0.2 przy maksymalnym powiększeniu makro ×8).
Przetwarzanie obrazu i analiza 3D
Przetwarzanie obrazu, segmentację i analizę zestawów danych arkuszy świetlnych przeprowadzono za pomocą ImageJ/Fiji, natomiast Figs. 3 i 4 zostały wygenerowane przy użyciu przeglądarki Imaris Viewer 9.9 (https://imaris.oxinst.com/imaris-viewer) i film uzupełniający 3 został wygenerowany za pomocą Imaris 9 (https://imaris.oxinst.com/) (Bitplane, Oxford Instruments). Kafelkowe zbiory danych w postaci arkuszy świetlnych zszyto za pomocą MosaicExplorerJ53. Segmentację 3D tkanki pęcherza moczowego przeprowadzono przy użyciu niestandardowych makr ImageJ/Fiji w celu półautomatycznej adnotacji 3D dużych objętości w trybie wirtualnym. W skrócie, pierwszy skrypt „Macro1” ładuje stosy obrazów 3D, umożliwia użytkownikowi dodawanie adnotacji do ROI w kilku płaszczyznach i automatycznie interpoluje ROI w celu wygenerowania i eksportowania masek 3D. ROI rysowano co 15 płaszczyzn (co 37.5 µm), aby zapewnić dobrą ciągłość segmentacji, przy jednoczesnym ograniczeniu adnotacji do rozsądnego minimum. Drugi skrypt, „Macro2”, wykonuje operacje matematyczne lub logiczne, płaszczyzna po płaszczyźnie, bez ładowania całych stosów do pamięci, pomiędzy maskami 3D lub pomiędzy maską 3D a oryginalnymi danymi, zapisując wynik jako nowy stos. Wszystkie maski wygenerowano poprzez dodanie adnotacji do obrazów autofluorescencji.
Zarówno warstwy powierzchniowe guza, jak i zdrowej tkanki (ryc. 3) zostały nakreślone za pomocą różdżki Fidżi i narzędzi lasso na jamie pęcherza w masce. Nazywając tę pierwszą iterację BC1, kolejne uruchomienia Macro1 automatycznie rozszerzają ten kontur 3D o określoną liczbę pikseli, aby uzyskać nowe iteracje maski, BC2, BC3 itd., wraz ze wzrostem rozszerzenia. Pierwszą warstwę zawierającą zarówno nowotwór, jak i zdrową tkankę, maskę L1, otrzymuje się poprzez odjęcie maski BC1 od BC2 itd., otrzymując L2 i L3 jako warstwy koncentryczne. Objętość guza najbliżej jamy uzyskano poprzez oznaczenie guza różdżką i narzędziami lasso w celu utworzenia maski T1, podczas gdy warstwę 3D zdrowego nabłonka dróg moczowych wykrywano oddzielnie w masce U1. Odejmując U1 od L1 otrzymujemy warstwę powierzchniową guza i tak dalej: L2 − U1, L3 − U1. I odwrotnie, pierwszą warstwę nabłonka dróg moczowych uzyskuje się poprzez odjęcie T1 od L1. Wszystkie warstwy na rys. 3 zdefiniowano jako grubość 33 µm.
Ten sam zestaw makr i procedur (narzędzie ImageJ różdżka, erozja cyfrowa 500 µm itd.) wykorzystano do nakreślenia i segmentacji wewnętrznej części tkanki pęcherza, a następnie oszacowania wewnętrznej objętości tkanki pęcherza (ryc. 4szczegóły powyżej). Histogramy intensywności rozproszonego sygnału utworzono na Fidżi poprzez połączenie rozproszonego sygnału i maski.
Używanie RNT 131Ja-nanoboty
Pomiędzy 8. a 15. dniem po wszczepieniu guza zwierzęta podzielono na sześć grup (grupy 1–6), starając się uzyskać podobną średnią objętość guza w poszczególnych grupach (tab. 2). Do eksperymentów zwierzęta poddano znieczuleniu (5% izofluran w czystym O2) i ułożyć się na wznak przed opróżnieniem pęcherza, masując okolicę brzucha. Bezpośrednio po tym 100 µl odpowiedniego preparatu o stężeniu 400 µg ml-1 (Stół 2) wkroplono do pęcherza przy użyciu cewnika o rozmiarze 24. Leczenie i nośnik (woda lub mocznik) pozostawały w pęcherzu przez 1 godzinę przed usunięciem cewnika. Pęcherz ponownie opróżniono poprzez masaż brzucha, a myszy wybudzono ze znieczulenia w swoich klatkach, zastępując trociny z klatek zwierzęcych 24 godziny po zabiegu w celu usunięcia skażenia radioaktywnego.
Skuteczność terapeutyczna określona za pomocą MRI
Na każdej myszy przeprowadzono dwa badania MRI: (1) pomiędzy 7 a 14 dniem po wszczepieniu nowotworu, w celu randomizacji zwierząt do grup i zmierzenia początkowej (przed leczeniem) objętości nowotworu; (2) pomiędzy 16 a 21 dniem po wszczepieniu guza (po leczeniu) w celu oceny skuteczności terapeutycznej. MRI przeprowadzono przy użyciu skanerów 7 T Bruker BioSpec i 11.7 T Bruker BioSpec (oba z oprogramowaniem ParaVision 7), w zależności od dostępności. Nie miało to wpływu na wyniki, ponieważ pole zewnętrzne nie jest krytyczne dla obrazowania anatomicznego14. Eksperymenty z obrazowaniem przeprowadzono przy użyciu tych samych parametrów obrazowania i przetwarzania, jak wyjaśniono powyżej (Śledzenie wielkości guza). W przypadku skanera 11.7 T układ składał się z cewki powierzchniowej serca myszy do odbioru i cewki wolumetrycznej do transmisji. Objętość guza w każdym skrawku określono na podstawie ręcznie pobranych objętości obejmujących obszar guza.
Analiza statystyczna
W badaniach obrazowych PET odsetek wstrzykniętej dawki (% ID) i wstrzykniętej dawki na objętość guza (% ID cm-3) porównano za pomocą jednoczynnikowej analizy ANOVA. Różnice między grupami określono za pomocą testu wielokrotnych porównań Tukeya. NTV w sekcji RNT uzyskano z a t-test wartości niesparowanych. Założono, że rozkład danych jest normalny, ale nie zostało to formalnie przetestowane. Analizy statystyczne przeprowadzono za pomocą programu GraphPad Prism v.8.
Podsumowanie raportowania
Dalsze informacje na temat projektu badań są dostępne w Podsumowanie sprawozdawczości dotyczącej portfela przyrody powiązane z tym artykułem.
- Dystrybucja treści i PR oparta na SEO. Uzyskaj wzmocnienie już dziś.
- PlatoData.Network Pionowe generatywne AI. Wzmocnij się. Dostęp tutaj.
- PlatoAiStream. Inteligencja Web3. Wiedza wzmocniona. Dostęp tutaj.
- PlatonESG. Węgiel Czysta technologia, Energia, Środowisko, Słoneczny, Gospodarowanie odpadami. Dostęp tutaj.
- Platon Zdrowie. Inteligencja w zakresie biotechnologii i badań klinicznych. Dostęp tutaj.
- Źródło: https://www.nature.com/articles/s41565-023-01577-y
- :Jest
- :nie
- ][P
- 000
- 1
- 10
- 100
- 11
- 13
- 14
- 15%
- 16
- 160
- 20
- 200
- 2015
- 2017
- 2018
- 2019
- 2020
- 2021
- 22
- 23
- 24
- 25
- 250
- 30
- 300
- 33
- 35%
- 3d
- 400
- 41
- 50
- 500
- 51
- 52
- 53
- 7
- 70
- 8
- 9
- 95%
- a
- O nas
- powyżej
- zgodność
- akumulacja
- Osiągać
- osiągnięty
- nabyty
- nabywanie
- nabycie
- w poprzek
- aktywowany
- aktywny
- działalność
- w dodatku
- dodanie
- do tego
- administracja
- oddziaływać
- Po
- potem
- ponownie
- wymierzony
- AIR
- AL
- alex
- algorytm
- Wszystkie kategorie
- dozwolony
- wzdłuż
- Alzheimera
- wśród
- ilość
- skrobiowaty
- an
- analizuje
- analiza
- Kotwica
- i
- zwierzę
- zwierzęta
- przeciwciało
- każdy
- aplikacje
- stosowany
- Stosowanie
- właściwy
- zatwierdzony
- Aqua
- SĄ
- POWIERZCHNIA
- artykuł
- AS
- przypuszczalny
- Założenia
- At
- Władze
- automatycznie
- dostępność
- dostępny
- średni
- średnio
- b
- BABB
- przepustowość
- podstawa
- BE
- zanim
- zachowanie
- jest
- poniżej
- pomiędzy
- Blokować
- ciało
- gotowany
- obie
- Mózg
- ciężar
- ale
- by
- klatki
- obliczony
- powołanie
- aparat fotograficzny
- Rak
- walizka
- Etui
- CCD
- komórka
- Komórki
- Zmiany
- opłata
- chemiczny
- Clearing
- kliknij
- Kliniczne
- Zamknij
- cewka
- połączenie
- łączenie
- handlowy
- komisja
- porównywalny
- w porównaniu
- porównania
- całkowicie
- wypełniając
- stężenie
- warunek
- Warunki
- przeprowadzone
- pewność siebie
- ZATWARDZIAŁY
- kolejny
- ciągłość
- bez przerwy
- kontrola
- kontrolowanych
- odwrotnie
- Chłodny
- poprawione
- Korekty
- Odpowiedni
- Rada
- sprzężony
- pokrycie
- Stwórz
- stworzony
- Tworzenie
- Twórczy
- Kryteria
- krytyczny
- Skany CT
- zwyczaj
- Ciąć
- Ciemny
- dane
- zbiory danych
- dzień
- Dni
- de
- zdefiniowane
- dostarczanie
- W zależności
- opisane
- Wnętrze
- życzenia
- detale
- wykryte
- Wykrywanie
- Ustalać
- ustalona
- określaniu
- rozwinięty
- urządzenie
- diagnostyka
- ZROBIŁ
- różnica
- Różnice
- różne
- Transmitowanie
- cyfrowy
- rozcieńczony
- kierunek
- choroba
- danie
- Dyspersja
- 分配
- Dystrybucje
- podzielony
- do
- robi
- zrobić
- Dawka
- sporządzony
- Spadek
- czas trwania
- dynamiczny
- dynamika
- e
- E i T
- każdy
- łatwo
- przegapić
- skuteczność
- bądź
- osadzone
- umożliwiać
- Umożliwia
- Punkt końcowy
- wzmocnione
- Cały
- enzymatyczny
- wyposażony
- ustanowiony
- ustanowienie
- oszacowanie
- Eter (ETH)
- etyka
- europejski
- oceniać
- Każdy
- badanie
- prace
- Z wyjątkiem
- oczekiwanie
- spodziewany
- eksperyment
- eksperymentalny
- eksperymenty
- wyjaśnione
- eksport
- zewnętrzny
- ułatwiać
- czynnik
- Płeć żeńska
- pole
- Figa
- Postać
- wypełniony
- filtrować
- filtry
- finał
- W końcu
- i terminów, a
- dopasowywanie
- ustalony
- Migać
- następnie
- następujący
- W razie zamówieenia projektu
- Nasz formularz
- formaldehyd
- Formalnie
- naprzód
- cztery
- od
- zamrożone
- funkcjonować
- dalej
- szczelina
- ge
- GE Healthcare
- Ogólne
- Generować
- wygenerowane
- Dający
- dobry
- gradienty
- gram
- Zielony
- Zarządzanie
- Grupy
- Wzrost
- wytyczne
- Have
- he
- Zdrowie
- opieki zdrowotnej
- zdrowy
- Serce
- HTTPS
- Huang
- człowiek
- icap
- ID
- identiques
- if
- obraz
- Analiza obrazu
- zdjęcia
- Obrazowanie
- natychmiast
- zanurzony
- poprawia
- in
- włączony
- włączenie
- wzrastający
- inkubowane
- Inkubacja
- inkubacja
- niezależny
- wskaźnik
- indukcja
- Informacja
- początkowy
- wewnętrzny
- wewnątrz
- instrumenty
- zintegrowany
- interaktywne
- odsetki
- wewnętrzny
- najnowszych
- wprowadzenie
- iteracja
- iteracje
- JEGO
- KDA
- konserwacja
- zestaw
- l2
- duży
- laser
- Lasery
- warstwa
- nioski
- pozostawiając
- lewo
- Obiektyw
- obiektywy
- życie
- lekki
- Linia
- LINK
- powiązany
- załadunek
- masa
- miejscowy
- od
- Macro
- makra
- utrzymać
- konserwacja
- ręcznie
- maska
- Maski
- dopasowywanie
- materiał
- materiały
- matematyczny
- Matrix
- maksymalny
- znaczy
- zmierzyć
- mierzona
- Pomiary
- zmierzenie
- Media
- Pamięć
- merynos
- Metanol
- metody
- Myszy
- Mikroskop
- Mikroskopia
- min
- minimum
- moll
- mieszany
- mieszanina
- ML
- Aplikacje mobilne
- Moda
- model
- modele
- zmodyfikowano
- moduł
- Cząsteczkowa
- monitorowane
- monitorowanie
- ruch
- mysz
- MRI
- MS
- MT
- wielokrotność
- nanotechnologia
- Natura
- sieci
- Neutralny
- Nowości
- Nie
- Hałas
- normalna
- cel
- zauważony
- uzyskać
- uzyskane
- of
- Stary
- on
- ONE
- operacyjny
- operacyjny
- operacje
- zoptymalizowane
- or
- Orca
- oryginalny
- koniec
- w ciągu nocy
- Oxford
- Tlen
- Papier
- parametry
- Park
- część
- cząstka
- PBS
- kołek
- penetracja
- dla
- wykonywane
- wykonuje
- zwierzę domowe
- Petri
- fazy
- Pivot
- piksel
- umieszczony
- samolot
- Planes
- plato
- Analiza danych Platona
- PlatoDane
- punkt
- zwrotnica
- teczka
- position
- ustawione
- Detaliczność
- przygotowany
- teraźniejszość
- poprzednio
- procedura
- procedury
- przetwarzanie
- stopniowo
- protokół
- Qi
- ilościowy
- radio
- przypadkowy
- Randomizowane
- SZCZUR
- Kurs
- stosunek
- React
- reakcja
- Czytelnik
- rozsądny
- Przyczyny
- Odebrane
- recepcja
- nagrany
- Recover
- Czerwony
- zmniejszyć
- Zredukowany
- zmniejsza
- odniesienie
- region
- pozostał
- szczątki
- usunąć
- Usunięto
- usuwanie
- Zgłoszone
- Raportowanie
- przedstawiciel
- wymagany
- Badania naukowe
- odporny
- Rozkład
- odpowiednio
- odpowiedź
- dalsze
- wynikły
- Efekt
- robot
- Pokój
- działa
- s
- SA
- taki sam
- oszczędność
- skany
- rozrzucone
- SCI
- naukowy
- scenariusz
- druga
- wtórny
- Sekcja
- działy
- widzieć
- segment
- segmentacja
- wybrany
- Sekwencja
- służył
- w panelu ustawień
- kilka
- arkusz
- pokazane
- Signal
- Dystrybucja sygnału
- podobny
- ponieważ
- pojedynczy
- SIX
- Rozmiar
- Plaster
- mały
- So
- sód
- Tworzenie
- rozwiązanie
- Rozwiązania
- Przestrzenne
- Stabilność
- stos
- Półki na książki
- statystyczny
- gwiezdny
- Cel
- przechowywany
- pas
- student
- badania naukowe
- kolejny
- Następnie
- apartament
- Powierzchnia
- objawy
- synteza
- system
- T
- T1
- stół
- biorąc
- tech
- Technologies
- teleskop
- szablon
- dziesięć
- test
- przetestowany
- że
- Połączenia
- ich
- Im
- następnie
- teoretyczny
- Terapeutyczny
- terapia
- to
- całkowicie
- tych
- trzy
- trójwymiarowy
- Przez
- poprzez
- czas
- czasy
- tkanka
- do
- razem
- narzędzie
- narzędzia
- Kwota produktów:
- w kierunku
- w kierunku
- transformatorowy
- leczenie
- Tryton
- stara
- guzy
- drugiej
- rodzaj
- zazwyczaj
- nieporuszony
- dla
- aż do
- na
- używany
- Użytkownik
- za pomocą
- Wartości
- pojazd
- Przeciw
- przez
- Wideo
- Filmy
- Zobacz i wysłuchaj
- Wirtualny
- wyobrażać sobie
- vivo
- Tom
- kłęby
- wolumetryczny
- chodzik
- różdżka
- była
- mycie
- Uzdatnianie wody
- we
- tygodni
- waga
- Dobrobyt
- były
- Koło
- który
- Podczas
- biały
- okna
- w
- w ciągu
- bez
- X
- Wydajność
- wydajność
- plony
- zefirnet
- zoom