La Food and Drug Administration (FDA) joue un rôle central dans la protection de la santé publique à travers la réglementation des dispositifs médicaux, garantissant qu'ils sont à la fois sûrs et efficaces pour l'usage auquel ils sont destinés. À l’époque contemporaine, l’essor des appareils connectés a placé la cybersécurité au premier plan des préoccupations de la FDA ; dans ce contexte, il est essentiel de définir des exigences spécifiques pour la définition du modèle de menace de cybersécurité. Alors que les dispositifs médicaux sont de plus en plus intégrés à la technologie, la nécessité de mesures de cybersécurité robustes pour protéger les informations des patients et garantir la fonctionnalité de ces appareils est plus critique que jamais. Le respect des réglementations FDA est non seulement essentiel pour la conformité des fabricants mais également pour la sécurité et la confiance des patients et des utilisateurs.
Nous avons discuté à plusieurs reprises des dispositifs médicaux numériques et des exigences associées sur les sites Web QualityMedDev, couvrant différents sujets tels que L'IA dans les dispositifs médicaux, produits de santé numériqueset une Approche TGA pour la régulation des dispositifs médicaux numériques. Parallèlement, la FDA a publié différentes lignes directrices en matière de cybersécurité, soit en matière de exigences préalables à la commercialisation ainsi que exigences post-commercialisation.
La FDA a établi des exigences en matière de cybersécurité dans le cadre de sa surveillance réglementaire afin de protéger les patients et de garantir l'intégrité des dispositifs médicaux. Ces normes s'appliquent à l'ensemble du cycle de vie d'un appareil, depuis la conception initiale et le développement jusqu'au déploiement et à la maintenance. Les dispositifs médicaux devenant de plus en plus intelligents et connectés, ils sont de plus en plus vulnérables aux cybermenaces. L'intégration des pratiques de cybersécurité pendant la phase de développement des dispositifs médicaux est une étape cruciale pour atténuer les risques posés par les cyberattaques.
Éléments clés du cadre de cybersécurité de la FDA
Pour garantir l'intégrité de la cybersécurité des dispositifs médicaux, la FDA a défini des exigences préalables à la commercialisation qui incluent la nécessité pour les fabricants d'intégrer un modèle de menace de cybersécurité et des contrôles de sécurité dès les premières étapes de la conception des dispositifs.
Les fabricants de dispositifs doivent établir et respecter des systèmes de qualité pour garantir une conformité constante aux exigences et spécifications applicables à leurs produits. Les exigences relatives à ces systèmes qualité peuvent être trouvées dans la réglementation du système qualité (QS) dans 21 CFR Part 820 si l'appareil est vendu aux États-Unis, ou dans d'autres réglementations pour d'autres pays (par exemple EU MDR 2017/745 pour l'Union européenne).
Selon la nature de l'appareil, les exigences QS peuvent être pertinentes pendant la phase de pré-commercialisation, la phase de post-commercialisation, ou les deux. Dans le contexte de la phase de pré-commercialisation, démontrer une assurance raisonnable de sécurité et d'efficacité pour certains appareils présentant des risques de cybersécurité peut impliquer d'inclure des documents relatifs au règlement QS dans le cadre de la soumission préalable à la commercialisation. Par exemple, les normes ISO 13485 :2016 et 21 CFR 820 exigent que les fabricants de toutes les classes de dispositifs automatisés avec un logiciel établissent des procédures pour contrôler la conception du dispositif, garantissant ainsi la conformité aux exigences de conception spécifiées (appelées « contrôles de conception »). Dans le cadre des contrôles de conception, les fabricants sont tenus « d'établir et de maintenir des procédures de validation de la conception du dispositif », qui englobent « la validation du logiciel et l'analyse des risques, le cas échéant » . Dans le cadre de la validation des logiciels et de l'analyse des risques obligatoires, les fabricants de dispositifs logiciels peuvent avoir besoin d'instituer des processus de gestion et de validation des risques de cybersécurité, le cas échéant.
La validation des logiciels et la gestion des risques constituent des éléments cruciaux des analyses de cybersécurité, déterminant si un appareil offre une assurance raisonnable de sécurité et d'efficacité. La FDA oblige les fabricants à intégrer des processus de développement qui prennent en compte et traitent les risques logiciels tout au long des étapes de conception et de développement, dans le cadre des contrôles de conception. Ces processus devraient englober des considérations de cybersécurité. Cela comprend l'identification des risques de sécurité, l'établissement d'exigences de conception pour contrôler ces risques et la fourniture de preuves que les contrôles fonctionnent comme prévu et sont efficaces dans l'environnement désigné de l'appareil, garantissant ainsi des mesures de sécurité suffisantes.
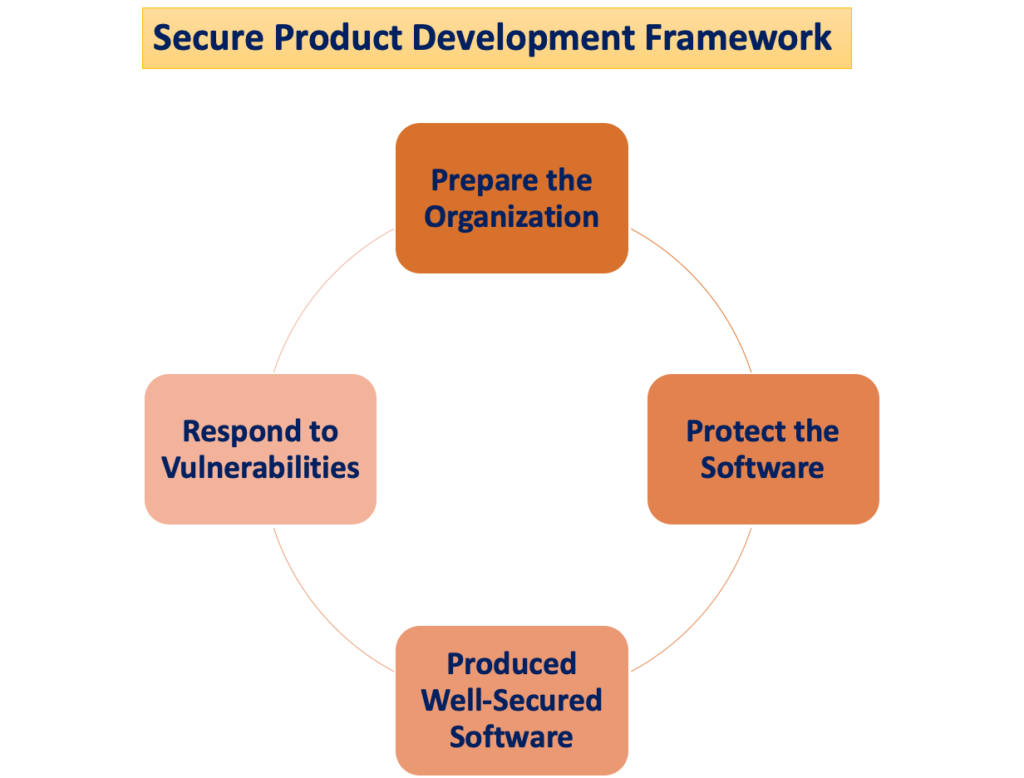
Le "Cadre de développement de produits sécurisé"
Le risque de préjudice pour les patients survient lorsque les menaces de cybersécurité exploitent les vulnérabilités d'un système, et la facilité avec laquelle ces menaces peuvent compromettre la sécurité et l'efficacité d'un dispositif médical augmente avec le nombre de vulnérabilités identifiées au fil du temps. Un cadre de développement de produits sécurisé (SPDF) comprend des processus visant à identifier et à réduire la quantité et la gravité des vulnérabilités des produits. Englobant toutes les étapes du cycle de vie d'un produit (conception, développement, publication, support et mise hors service), le SPDF fait partie intégrante.
Lors de la conception des appareils, l'intégration des processus SPDF peut éviter la nécessité d'une réingénierie lors de l'intégration de fonctionnalités basées sur la connectivité après commercialisation ou de la résolution de vulnérabilités qui posent des risques incontrôlés. L'intégration réalisable avec les processus existants pour le développement de produits et de logiciels, la gestion des risques et le système qualité plus large ajoute à la polyvalence du SPDF.
Pour garantir le respect de la réglementation Système Qualité (QS), l’utilisation d’un SPDF est recommandée. La FDA encourage les fabricants à adopter le SPDF pour ses avantages en matière de respect de la réglementation QS et des exigences de cybersécurité. Il est toutefois reconnu que d'autres approches peuvent également satisfaire à la réglementation QS.
Gestion des risques de cybersécurité
L’objectif principal de l’utilisation d’un SPDF (Secure Product Development Framework) est de créer et de maintenir des appareils à la fois sûrs et efficaces. Du point de vue de la sécurité, ces appareils gagnent également en fiabilité et en résilience. Les fabricants et/ou les utilisateurs (par exemple, les patients, les établissements de santé) peuvent ensuite gérer ces appareils, y compris l'installation, la configuration, les mises à jour et l'examen des journaux des appareils, via la conception des appareils et l'étiquetage associé.
Les établissements de santé ont la possibilité de gérer ces appareils dans le cadre de leurs propres cadres de gestion des risques de cybersécurité, tels que le National Institute of Standards and Technology, largement reconnu (NIST) Cadre pour l'amélioration de la cybersécurité des infrastructures critiques, communément appelé le Cadre de cybersécurité du NIST ou NIST CSF.
La FDA recommande aux fabricants d'intégrer des processus de conception d'appareils, tels que ceux décrits dans la réglementation du système qualité (QS), pour renforcer la sécurité du développement et de la maintenance des produits. Tout en préservant la flexibilité pour les fabricants, ils peuvent également explorer des cadres alternatifs qui s'alignent sur la réglementation QS et adhèrent aux recommandations de la FDA pour la mise en œuvre d'un SPDF. Les exemples incluent le cadre spécifique aux dispositifs médicaux dans le plan commun de sécurité des dispositifs médicaux et des technologies de l'information de santé (JSP) 30 et IEC 81001-5-1. Les cadres d'autres secteurs, tels que la norme ANSI/ISA 62443-4-1 Sécurité pour les systèmes d'automatisation et de contrôle industriels Partie 4-1 : Exigences du cycle de vie du développement de la sécurité des produits, peuvent également répondre aux réglementations QS.
Dans les sections suivantes de cet article, des recommandations pour l'utilisation des processus SPDF sont fournies, telles que généralement perçues par la FDA, qui mettent en évidence des considérations cruciales pour le développement de dispositifs sûrs et efficaces. Ces processus complètent la réglementation QS et la FDA suggère aux fabricants d'inclure la documentation correspondante pour examen dans les soumissions préalables à la commercialisation.
Modèle de menace de cybersécurité
La modélisation des menaces implique un processus systématique d'identification des objectifs de sécurité, des risques et des vulnérabilités dans l'ensemble du système de dispositifs médicaux. Par la suite, cela implique de définir des contre-mesures pour prévenir, atténuer, surveiller ou répondre aux impacts des menaces tout au long du cycle de vie du système de dispositif médical. Lorsqu'elle est appliquée de manière appropriée et complète, elle sert de base à l'optimisation de la sécurité des composants du système, des produits, des réseaux, des applications et des connexions.
Concernant la gestion des risques de sécurité et pour identifier les risques et contrôles de sécurité appropriés pour le système de dispositifs médicaux, la FDA préconise la mise en œuvre d'une modélisation des menaces pour éclairer et soutenir les activités d'analyse des risques. Dans le contexte de l’évaluation des risques, la FDA suggère d’intégrer la modélisation des menaces tout au long du processus de conception, englobant tous les éléments du système des dispositifs médicaux.
Les aspects clés du modèle de menace devraient englober l’identification des risques et les mesures d’atténuation, éclairant à la fois les risques avant et après l’atténuation pris en compte dans l’évaluation des risques de cybersécurité. De plus, le modèle doit formuler des hypothèses sur le système de dispositifs médicaux ou sur l’environnement d’utilisation, par exemple en supposant que les réseaux hospitaliers sont intrinsèquement hostiles. Cette hypothèse incite les fabricants à envisager des scénarios dans lesquels un adversaire contrôle le réseau, capable de modifier, d'abandonner et de rejouer des paquets. En outre, le modèle de menace doit capturer les risques de cybersécurité introduits par la chaîne d'approvisionnement, la fabrication, le déploiement, l'interopérabilité avec d'autres appareils, les activités de maintenance/mise à jour et les activités de déclassement, qui pourraient être négligés dans un processus traditionnel d'évaluation des risques de sécurité.
Pour les soumissions préalables à la commercialisation, la FDA recommande d'inclure une documentation de modélisation des menaces pour présenter l'analyse du système de dispositif médical, identifiant les risques de sécurité potentiels qui pourraient avoir un impact sur la sécurité et l'efficacité. Les fabricants ont la possibilité de choisir parmi diverses méthodologies ou combinaisons de méthodes pour la modélisation des menaces, et la justification de leur choix de méthodologie doit être fournie avec la documentation.
Il est conseillé de mener des activités de modélisation des menaces lors des revues de conception, et la documentation doit fournir suffisamment d'informations pour que la FDA puisse évaluer et examiner les fonctionnalités de sécurité intégrées dans l'appareil. Cette évaluation globale doit prendre en compte à la fois le dispositif et le système plus large dans lequel il fonctionne, en mettant l'accent sur la sécurité et l'efficacité du dispositif.
Les principaux éléments des modèles de menace de cybersécurité peuvent être résumés comme suit :
Identification des menaces potentielles
Le développement d'un modèle de menace constitue une étape fondamentale du processus de cybersécurité, permettant aux fabricants d'identifier et de comprendre les menaces potentielles de cybersécurité spécifiques à leurs dispositifs médicaux. Le développement d'un modèle de menace constitue une étape fondamentale du processus de cybersécurité, permettant aux fabricants d'identifier et de comprendre les menaces potentielles de cybersécurité spécifiques à leurs dispositifs médicaux. Le développement d'un modèle de menace constitue une étape fondamentale du processus de cybersécurité, permettant aux fabricants d'identifier et de comprendre les menaces potentielles de cybersécurité spécifiques à leurs dispositifs médicaux.
Évaluation et gestion des risques
L'évaluation et la gestion des risques impliquent l'évaluation des menaces identifiées pour établir leur impact potentiel et la conception de stratégies appropriées pour atténuer efficacement ces risques.
Mise en œuvre des contrôles de sécurité
Les contrôles de sécurité sont les garanties ou contre-mesures mises en œuvre pour protéger la confidentialité, l'intégrité et la disponibilité du dispositif médical contre les menaces identifiées.
Tendances futures des directives de cybersécurité de la FDA
À mesure que les menaces et la technologie évoluent, les directives de cybersécurité de la FDA évolueront également. Il est essentiel pour les industriels de rester informés et agiles face à ces changements. Les fabricants doivent anticiper les mises à jour des réglementations en se tenant au courant des tendances du secteur et en maintenant une approche proactive en matière de cybersécurité.
La préparation aux défis imprévus est essentielle ; l’établissement de protocoles de sécurité robustes et de stratégies tournées vers l’avenir permettra aux fabricants de répondre efficacement à l’état futur des réglementations en matière de cybersécurité.
La cybersécurité est un aspect de la réglementation des dispositifs médicaux qui ne peut être surestimé : elle est aussi intrinsèque à la sécurité des dispositifs que n'importe quelle fonctionnalité mécanique ou électrique. La FDA l’a reconnu et en mettant en œuvre un cadre complet de cybersécurité, elle vise à suivre le rythme de l’innovation tout en protégeant la santé publique. Les fabricants, les professionnels de santé et les patients doivent tous collaborer pour respecter ces normes et garantir la fiabilité et la sécurité continues des dispositifs médicaux face aux cybermenaces.
Abonnez-vous à la newsletter QualityMedDev
QualityMedDev est une plate-forme en ligne axée sur les sujets de qualité et de réglementation pour le secteur des dispositifs médicaux ; Suivez-nous sur LinkedIn ainsi que Twitter pour rester à jour avec les nouvelles les plus importantes sur le domaine de la réglementation.
QualityMedDev est l'une des plus grandes plateformes en ligne prenant en charge le secteur des dispositifs médicaux pour les sujets de conformité réglementaire. Nous fournissons services de conseil en réglementation sur un large éventail de sujets, de UE MDR & IVDR à ISO 13485, y compris la gestion des risques, la biocompatibilité, la convivialité et la vérification et la validation des logiciels et, en général, l'assistance à la préparation de la documentation technique pour le MDR.
Notre plateforme sœur Académie QualitéMedDev offre la possibilité de suivre des cours de formation en ligne et à votre rythme axés sur des sujets de conformité réglementaire pour les dispositifs médicaux. Ces formations, développées en collaboration avec des professionnels hautement qualifiés du secteur des dispositifs médicaux, vous permettent d'augmenter de manière exponentielle vos compétences sur un large éventail de sujets de qualité et de réglementation pour les opérations commerciales des dispositifs médicaux.

N'hésitez pas à vous inscrire à notre Newsletter !
- Contenu propulsé par le référencement et distribution de relations publiques. Soyez amplifié aujourd'hui.
- PlatoData.Network Ai générative verticale. Autonomisez-vous. Accéder ici.
- PlatoAiStream. Intelligence Web3. Connaissance Amplifiée. Accéder ici.
- PlatonESG. Carbone, Technologie propre, Énergie, Environnement, Solaire, La gestion des déchets. Accéder ici.
- PlatoHealth. Veille biotechnologique et essais cliniques. Accéder ici.
- La source: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- :possède
- :est
- :ne pas
- :où
- $UP
- 2016
- 30
- 350
- 820
- 9
- a
- A Propos
- Académie
- reconnu
- à travers
- d'activités
- En outre
- propos
- adresser
- Ajoute
- adhérer
- administration
- informé
- défenseurs
- agile
- Destinée
- vise
- aligner
- Tous
- permet
- aussi
- alternative
- an
- analyses
- selon une analyse de l’Université de Princeton
- ainsi que
- anticiper
- tous
- en vigueur
- applications
- appliqué
- Appliquer
- une approche
- approches
- approprié
- de manière appropriée
- SONT
- article
- AS
- d'aspect
- aspects
- Evaluer
- Évaluation de risque climatique
- associé
- hypothèse
- hypothèses
- assurance
- At
- Attaques
- Automatisation
- Automation
- disponibilité
- BE
- devenez
- devenir
- était
- avantages.
- traversin
- tous les deux
- vaste
- plus large
- Apporté
- la performance des entreprises
- opérations commerciales
- mais
- by
- CAN
- ne peut pas
- capable
- capturer
- certaines
- chaîne
- globaux
- Modifications
- Selectionnez
- choisi
- les classes
- collaborons
- collaboration
- COM
- комбинации
- communément
- Complément
- conformité
- composants électriques
- complet
- compromis
- conception
- Préoccupations
- confidentialité
- configuration
- connecté
- appareils connectés
- Connexions
- Considérer
- considérations
- considéré
- cohérent
- consiste
- constituer
- consulting
- contemporain
- contexte
- a continué
- des bactéries
- contrôle
- contrôles
- Correspondant
- pourriez
- d'exportation
- cours
- couvrant
- engendrent
- critique
- Infrastructure critique
- crucial
- cyber
- Cyber-attaques
- Cybersécurité
- Date
- définir
- définition
- démontrer
- déploiement
- Conception
- procédé de design
- désigné
- détermination
- développé
- développement
- Développement
- dispositif
- Compatibles
- différent
- numérique
- diminuer
- discuter
- Documentation
- Goutte
- drogue
- pendant
- e
- le plus tôt
- gagner
- facilité
- Efficace
- de manière efficace
- efficacité
- non plus
- éléments
- embrasser
- mettant l'accent
- employant
- permettant
- englober
- englobe
- englobant
- encourage
- assurer
- assurer
- Environment
- essential
- établir
- établies
- établissement
- Ether (ETH)
- EU
- du
- Union européenne
- évaluer
- évaluation
- JAMAIS
- preuve
- évolue
- exemples
- existant
- Exploiter
- explorez
- exponentielle
- Visage
- installations
- fda
- réalisable
- Fonctionnalité
- Fonctionnalités:
- champ
- Flexibilité
- concentré
- suivre
- Abonnement
- suit
- nourriture
- Administration des aliments et des médicaments
- Administration des aliments et des médicaments (FDA)
- Pour
- Premier plan
- tourné vers l'avenir
- trouvé
- Fondation
- Fondatrice
- Framework
- cadres
- De
- plein
- En outre
- avenir
- Général
- généralement
- lignes directrices
- nuire
- Vous avez
- Santé
- la médecine
- Haute
- Souligner
- très
- holistique
- Hôpital
- Cependant
- HTML
- HTTPS
- Identification
- identifié
- identifier
- identifier
- if
- Impact
- Impacts
- la mise en oeuvre
- mis en œuvre
- la mise en œuvre
- important
- l'amélioration de
- in
- comprendre
- inclut
- Y compris
- intégrer
- incorporation
- Améliore
- Augmente
- de plus en plus
- industriel
- l'automatisation industrielle
- industrie
- informer
- d'information
- Actualités
- Infrastructure
- intrinsèquement
- initiale
- Innovation
- installation
- instance
- Institut
- intégrale
- des services
- Intégration
- l'intégration
- intégrité
- prévu
- développement
- intrinsèque
- introduit
- impliquer
- implique
- ISO
- IT
- SES
- joint
- XNUMX éléments à
- ACTIVITES
- l'étiquetage
- le plus grand
- cycle de la vie
- vos produits
- MailChimp
- Entrée
- maintenir
- le maintien
- facile
- gérer
- gestion
- mandats
- Fabricants
- fabrication
- largeur maximale
- Mai..
- MDR
- les mesures
- mécanique
- médical
- dispositif médical
- dispositifs médicaux
- Découvrez
- réunion
- méthodologies
- méthodes
- pourrait
- Réduire les
- atténuer
- atténuer les risques
- modèle
- modélisation statistique
- numériques jumeaux (digital twin models)
- Surveiller
- PLUS
- (en fait, presque toutes)
- must
- Nationales
- Nature
- Navigation
- une nécessité
- Besoin
- réseau et
- réseaux
- nouvelles
- nist
- nombre
- objectif
- objectifs
- of
- on
- ONE
- en ligne
- uniquement
- fonctionner
- exploite
- Opérations
- l'optimisation
- Option
- or
- Autre
- nos
- décrit
- sorties
- plus de
- Surveillance
- propre
- Rythme
- paquets
- partie
- patientforward
- patients
- perçu
- objectifs
- phase
- pivot
- plan
- plateforme
- Platon
- Intelligence des données Platon
- PlatonDonnées
- joue
- plug-in
- posé
- position
- possibilité
- Poteaux
- défaillances
- pratiques
- préparation
- empêcher
- primaire
- Cybersécurité
- procédures
- processus
- les process
- Produit
- le développement de produits
- Produits
- ,une équipe de professionnels qualifiés
- instructions
- protéger
- L'utilisation de sélénite dans un espace est un excellent moyen de neutraliser l'énergie instable ou négative.
- protocoles
- fournir
- à condition de
- fournit
- aportando
- public
- santé publique
- publié
- qualité
- Quantité
- gamme
- raisonnement
- raisonnable
- reconnu
- recommandations
- recommandé
- recommande
- visée
- Règlement
- règlements
- régulateurs
- Conformité réglementaire
- en relation
- rapport
- libérer
- fiabilité
- rester
- conditions
- Exigences
- la résilience
- Réagir
- Avis
- Avis
- Augmenter
- Analyse
- évaluation des risques
- la gestion des risques
- risques
- robuste
- Rôle
- des
- la sauvegarde
- garanties
- Sécurité
- même
- scénarios
- les sections
- secteur
- Secteurs
- sécurisé
- sécurité
- Mesures de sécurité
- gestion des risques de sécurité
- risques de sécurité
- sert
- plusieurs
- gravité
- devrait
- vitrine
- sœur
- qualifié
- plus intelligents
- So
- Logiciels
- développement de logiciels
- vendu
- groupe de neurones
- caractéristiques
- spécifié
- Étape
- étapes
- Normes
- Région
- États
- rester
- rester
- étapes
- les stratégies
- Soumission
- Soumission
- inscrire
- Par la suite
- tel
- suffisant
- Suggère
- convient
- la quantité
- chaîne d'approvisionnement
- Support
- Appuyer
- combustion propre
- Système
- Technique
- Technologie
- que
- qui
- La
- El futuro
- leur
- puis
- Ces
- l'ont
- this
- ceux
- menace
- des menaces
- Avec
- tout au long de
- fiable
- fois
- à
- trop
- Les sujets
- traditionnel
- Formation
- Trends
- La confiance
- fiabilité
- comprendre
- imprévu
- union
- Uni
- États-Unis
- Actualités
- Défendre
- URL
- us
- convivialité
- Utilisation
- utilisé
- utilisateurs
- Utilisant
- valider
- validation
- divers
- Vérification
- la versatilité
- vulnérabilités
- Vulnérable
- we
- sites Internet
- quand
- que
- qui
- tout en
- largement
- sera
- comprenant
- dans les
- Outils de gestion
- Plugin WordPress
- you
- Votre
- zéphyrnet





