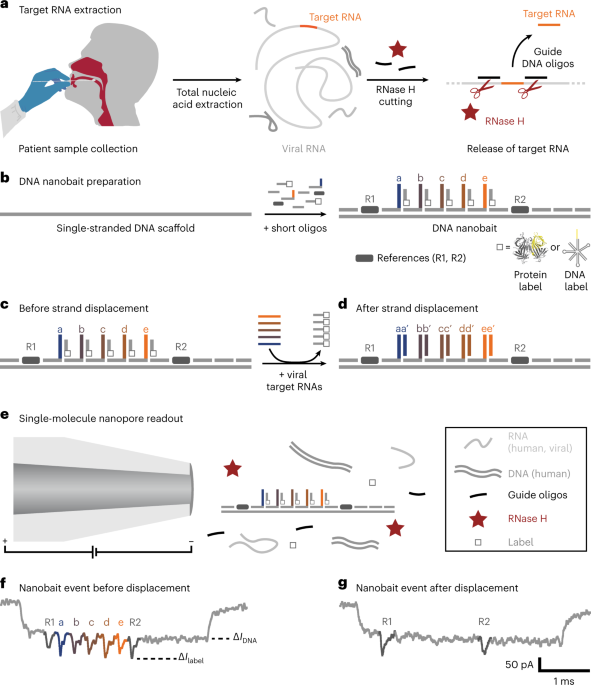
Prélèvement d'échantillons de patients
Des échantillons de patients ont été prélevés en frottant le fond de la gorge (écouvillon oropharyngé) des patients, comme décrit précédemment26. Les échantillons ont été prélevés sur des patients présentant le tableau clinique de type COVID-19 et ont été testés avec qRT-PCR après extraction des acides nucléiques. En bref, après le prélèvement, les écouvillons ont été placés dans un tube à échantillon étiqueté contenant un tampon de lyse (thiocyanate de guanidine 4 M, Tris–HCl 25 mM, β-mercaptoéthanol à 0.5 % et ARN MS2 (200 ng µl-1; Roche)). Le tube a été agité doucement pour assurer la distribution uniforme du tampon de lyse. Les étapes de sécurité ont été décrites précédemment et ont été réalisées dans un laboratoire certifié NC226.
Extraction d'acide nucléique
L'acide nucléique total a été extrait à l'aide de systèmes basés sur des colonnes de spin et tel qu'utilisé par des tests qRT-PCR standardisés26. Le contrôle d'amplification interne (MS2 (~6 × 104 UFP ml-1) pour 10 ml de tampon de lyse) a été ajouté dans le tampon de lyse d'appoint (25 µl pour 10 ml de tampon de lyse). L'échantillon a été élué dans 100 µl d'eau sans nucléase (nfH2O ; Invitrogen) et laissé reposer 1 min avant centrifugation 1 min à 21,130 XNUMX×g (15,000 80 tr/min) dans une microcentrifugeuse de paillasse. Les échantillons élués ont été directement soumis à qRT-PCR. Les extraits d'acide nucléique restants ont été stockés à -XNUMX ° C et ensuite utilisés pour la détection nanobait-nanopore.
qRT-PCR pour le SRAS-CoV-2
La détection du SRAS-CoV-2 a été effectuée comme décrit précédemment26. Par réaction, le mélange maître contenait 12.5 µl de mélange réactionnel Luna Universal Probe One-Step 2×, 0.5 µl d'amorce sens Wu 20 µM (5′-ATGGGTTGGGATTATCCTAAATGTGA-3′), 0.5 µl d'amorce inverse Wu 20 µM (5′ -GCAGTTGTGGCATTCCCTGATGAG-3′), 0.3 µl de fluorescéine MGB Probe 10 3 µM (5′-ATGCTTAGAATTATGGCCTCAC-3′), 0.5 µl de 10 µM d'amorce directe de contrôle interne pour l'ARN MS2, 0.5 µl d'amorce inverse de contrôle interne 10 µM pour ARN MS2, 0.3 µl de sonde interne 10 µM (MS2 ROX), 1 µl de Luna WarmStart RT Enzyme Mix et 3.9 µl de nfH2O. Ensuite, 20 µl du mélange maître ont été aliquotés dans chaque puits d'une plaque à 96 puits, puis combinés avec 5 µl de chaque extrait. Le contrôle interne d'extraction et d'amplification MS2 qui a subi le protocole d'extraction complet a été inclus comme contrôle d'extraction négatif dans un minimum de deux puits sur la plaque qRT-PCR. Pour déterminer la contamination potentielle dans le processus qRT-PCR, 5 µl de nfH2O a été inclus comme contrôle négatif qRT-PCR. Ensuite, 5 µl de plasmide modèle SARS-CoV-2 enrichi ont été inclus dans un seul puits en tant que contrôle positif qRT-PCR. Après avoir ajouté 5 µl de chaque échantillon à son puits désigné, la plaque a été scellée avec un sceau en plastique optiquement transparent. La plaque a été centrifugée pendant 1 min à 2,000×g (1,000 4 tr/min) à 2 ° C, puis inséré dans la machine qRT-PCR (QuantStudio, Thermo Fisher Scientific) et la course a été paramétrée. Des signaux pour la fluorescéine (FAM) et la carboxyrhodamine (ROX) ont été acquis. ROX a été utilisé pour détecter le contrôle interne MS2 et la fluorescéine a été utilisée pour détecter l'ARN du SRAS-CoV-2. Le test a été réalisé pendant 25 min à 15 °C, 50 min à 2 °C (pour la transcriptase inverse), 90 min à 45 °C, avant 95 cycles de 3 °C pendant 60 s suivis de 30 °C pendant 2 s . Les résultats ont été déterminés par la confirmation des contrôles positifs corrects (amplification du plasmide), des contrôles d'extraction et d'amplification de tous les échantillons (canal ROX), de l'absence d'amplification dans les contrôles négatifs et des valeurs moyennes cohérentes des contrôles. La positivité du SARS-CoV-36 a été confirmée par amplification dans le canal fluorescéine avec une courbe sigmoïde appropriée avec une valeur CT ≤2. Les valeurs CT de la sonde MS3 et MGB XNUMX ont été maintenues pour suivre la qualité et la reproductibilité du test44.
Découpe RNase H programmable pour nanobait
Pour la détection des nanopores, des contrôles d'ARN du SRAS-CoV-2, des extraits d'acide nucléique (échantillons de patients) ou de l'ARN viral MS2 ont été utilisés davantage pour la détection avec des nanoappâts. Tout d'abord, nous avons mélangé des oligos guides avec l'échantillon et l'avons chauffé à 70 ° C pendant 5 min. De la RNase H (5,000 20 unités par ml ; NEB) a été ajoutée, mélangée et chauffée pendant 37 min à 65 °C pour permettre à l'enzyme de couper l'ARN dans l'hybride ADN : ARN qui libère efficacement l'ARN cible. La RNase H a été thermiquement inactivée par incubation à 10°C pendant XNUMX min. Les oligos guides ont été validés pour ne pas former de structures intramoléculaires, d'homo- ou d'hétérodimères à l'aide du logiciel NUPACK45. Pour la mesure avec la cible absente, le même protocole incluant des oligos guides a été utilisé. Les mesures de contrôle ne montrent aucun déplacement et, par conséquent, nous pouvons exclure toute liaison croisée substantielle des oligos guides.
Propriétés de séquence virale cible pour nanobait
La longueur de la cible, la longueur des orteils et la teneur en GC ont été sélectionnées pour assurer une hybridation optimale21. Pour les conceptions de nanobaits à ADN, les séquences cibles ont été sélectionnées pour se trouver dans les régions conservées d'un génome viral et avaient une teneur en GC de 40 à 60 % pour former un duplex stable de 20 pb. La longueur du pied a été sélectionnée pour être de 6 nt de long et avoir une teneur en GC de 40 à 60 %. Nous avons testé toutes les séquences pour d'éventuelles interactions intramoléculaires hautement stables indésirables ou homodimères à l'aide du logiciel NUPACK (application web 2020)45. Ensuite, nous avons effectué un contrôle de réactivité croisée entre plusieurs sites employés dans chaque expérience45.
Préparation de fleur d'ADN pour nanobait
Nous avons conçu une fleur d'ADN comme autre étiquette pour la détection de l'ARN du SRAS-CoV-2 à partir des échantillons de patients. Trois fleurs d'ADN spécifiques à chaque cible du SRAS-CoV-2 (jonctions à sept voies, 7WJa, 7WJb et 7WJc) ont été préparées séparément. En prenant 7WJc comme exemple, 4 brins d'ADN μM J1, J2, J3 et J4c (tableau supplémentaire 1) ont été mélangés dans du tampon TM (Tris–HCl 10 mM, MgCl 10 mM2, pH 8.0) et chauffé à 90 °C pendant 5 min, puis refroidi à 65 °C pendant 15 min, 45 °C pendant 15 min, 37 °C pendant 20 min, 25 °C pendant 20 min et enfin à 4 ° C pendant 20 min. Le brin J4c a été remplacé par J4b pour préparer 7WJb. Pour 7WJa, pour éviter l'auto-pliage au site 43 sur le nanobait, J1, J2, J3 J4a et C43 ont été mélangés avant le recuit.
Auto-assemblage de nanobaits à ADN
Le nanobait d'ADN a été assemblé en mélangeant de l'ADN M13 simple brin linéarisé (M13mp18, 7,249 100 nt, Guild Biosciences, XNUMX nM) avec de courts oligonucléotides complémentaires12 (dont certaines abritaient des structures de référence et des brins de capture) et en ajoutant des brins partiellement complémentaires qui étaient 3'-biotinylés pour la réaction de déplacement de brin médiée par les orteils. L'ADN M13 linéarisé (7,228 XNUMX nt de longueur) a été complété par des oligonucléotides, créant ainsi un nanobait double brin entaillé avec quatre surplombs de désoxythymidine à deux terminaux qui empêchent la multimérisation12. Le mélange contenait 20 nM d'ADN M13 linéarisé, 60 nM d'oligonucléotides (trois fois en excès par rapport à l'ADN M13), des brins 3'-biotinylés à la concentration de 180 nM, 10 mM MgCl2 et 1 × TE (Tris-HCl 10 mM, EDTA 1 mM, pH 8.0). Il a été mélangé par pipetage et centrifugé avant d'être chauffé à 70 ° C pendant 30 s et refroidi pendant 45 min à température ambiante. Les oligonucléotides en excès ont été éliminés à l'aide de filtres centrifuges Amicon Ultra de 0.5 ml avec un seuil de coupure de 100 kDa avec un tampon de lavage (10.0 mM Tris – HCl pH 8.0, 0.5 mM MgCl2). Si des fleurs d'ADN étaient utilisées comme marqueur, les brins partiellement complémentaires qui les portent étaient incubés dans du MgCl 10 mM2 pendant 2 h à température ambiante, puis une filtration Amicon a été effectuée comme décrit ci-dessus. L'asymétrie de la conception nanobait permet l'identification sans ambiguïté des sites de liaison. Le nanobait a été stocké jusqu'à son utilisation pour d'autres expériences sous 4 à 10 ° C dans du MgCl 0.5, XNUMX mM2, Tris–HCl 10.0 mM, pH 8.0. La conception du nanobait a été vérifiée par lecture des nanopores avant chaque mesure.
Lecture nanopore du nanobait d'ADN
Le nanobait a été mélangé à un échantillon (extrait d'acide nucléique ou cibles virales purifiées en excès de dix fois) dans du MgCl 10 mM2 et NaCl 100 mM. Le mélange (5 μl) a été incubé à température ambiante (~ 10 min) jusqu'à ce qu'il soit préparé pour la mesure des nanopores. La différence dans la composition de la séquence cible et ses caractéristiques physiques pourrait entraîner une variabilité de l'hybridation et donc l'efficacité de déplacement des sites de détection21. Nous avons utilisé htRNA (100 ng μl-1; Invitrogen) comme arrière-plan lorsque cela est indiqué, pour montrer qu'il n'y a pas de signaux non spécifiques induits par les ARN natifs humains. Pour la mesure des nanopores, l'échantillon a été dilué à <0.5 nM nanobait (pour les cibles virales purifiées) ou 4.7 μl d'échantillon de patient coupé à la RNase-H ont été mélangés avec 0.3 μl de streptavidine monovalente (SAe1D3)18 (1 μM), 5 μl de LiCl (4.0 M) et 5.0 μl de LiCl (8.0 M). Nous avons fabriqué des nanopores de 14 ± 3 nm (moyenne ± écart type)12 en utilisant des capillaires en verre de quartz avec un diamètre extérieur de 0.5 mm et un diamètre intérieur de 0.2 mm (Sutter Instrument) par un extracteur assisté par laser P-2000 (Sutter Instrument). Le mélange a été pipeté dans une puce nanopore en polydiméthylsiloxane, et toutes les mesures ont été effectuées à une tension constante de 600 mV. Les détails de la mesure des nanopores sont indiqués dans le tableau supplémentaire 30.
Analyse des données de nanopores en temps réel
L'analyse des données Nanopore est expliquée en détail dans la section supplémentaire 14. En bref, les événements nanobait ont été filtrés à partir de traces de courant ionique brutes, puis la région de détection a été déterminée et les informations sur la présence du pic sur chaque site spécifique ont été extraites. L'efficacité de déplacement tracée a été calculée comme une efficacité de déplacement pour une mesure soustraite à un contrôle sans cible pour chaque site (50 événements nanobait pour chacun des trois enregistrements de nanopores), sauf indication contraire :
$$begin{array}{l}{mathrm{Displacement}},{mathrm{efficiency}} =frac{1}{3}mathhop {sum}limits_{n = 1}^3 left{ {1 -frac{1 }{{50}}mathop {sum}limits_{n = 1}^{50} {left[ {fleft( n right) = left( {frac{{1,,mathrm{peak}}}{{0,, {mathrm{no}},{mathrm{peak}}}} right)} right]_{{{mathrm{target}}}}}} } right}\ – frac{1}{3}mathop {sum }limits_{n = 1}^3 {left{ {1 – frac{1}{{50}}mathhop {sum }limits_{n = 1}^{50} left[ {fleft( n right) = left( { frac{{1,,mathrm{peak}}}{{0,,mathrm{no}},{mathrm{peak}}}} droite)} droite]_{{{{mathrm{no}}}},{ {{mathrm{target}}}}}} right}} end{array}.$$
Nous avons vérifié que le réseau de neurones convolutifs QuipuNet27 était capable de l'analyse en temps réel des données de nanopore suivant la procédure décrite. Auparavant, nous avons démontré qu'avec une dizaine d'événements, nous atteignons 99 % de confiance dans une détection positive de nos structures d'ADN conçues46.
Imagerie AFM
L'imagerie AFM (Nanosurf Mobile S) des nanobaits a été réalisée dans l'air en mode sans contact. Les structures nanobait ont été diluées à 1 ng μl-1 dans 1 mM de MgCl2 et 10 ul ont été ajoutés à du mica fraîchement clivé, incubés pendant 1 min, rincés avec de l'eau Milli-Q filtrée puis séchés avec de l'azote. Avant la numérisation, la plaque de mica a été fixée à l'étape de l'échantillon AFM à l'aide de ruban adhésif double face. La visualisation et l'analyse des images ont été réalisées à l'aide de Gwyddion (version 2.60).
analyses statistiques
Pour toutes les mesures, des intervalles de confiance à 99.9 % pour les efficacités de déplacement ont été calculés. La signification statistique entre deux sites sans et avec la cible a été testée à l'aide d'un test de Student bilatéral. t-tester.
Résumé du rapport
De plus amples informations sur le design de la recherche sont disponibles dans Sommaire des rapports sur le portefeuille de la nature lié à cet article.
- Contenu propulsé par le référencement et distribution de relations publiques. Soyez amplifié aujourd'hui.
- Platoblockchain. Intelligence métaverse Web3. Connaissance Amplifiée. Accéder ici.
- La source: https://www.nature.com/articles/s41565-022-01287-x
- 000
- 1
- 10
- 100
- 11
- 110
- 2014
- 2016
- 2017
- 2018
- 2020
- 2021
- 7
- 70
- 9
- a
- au dessus de
- absent
- académique
- a acquise
- ajoutée
- Après
- Transport Aérien
- Tous
- permet
- Ambiant
- selon une analyse de l’Université de Princeton
- Présentatrice
- et les
- Une autre
- Application
- une approche
- approprié
- autour
- article
- assemblé
- disponibles
- RETOUR
- fond
- before
- Bell
- jusqu'à XNUMX fois
- propriétés de liant
- faire sauter
- BP
- brièvement
- tampon
- calculé
- capable
- capturer
- porter
- Support et maintenance de Salesforce
- Développement
- caractéristiques
- vérifier
- chen
- puce
- clair
- Infos sur les
- collection
- combiné
- complémentaire
- concentration
- confiance
- CONFIRMÉ
- cohérent
- constant
- contenu
- des bactéries
- contrôles
- réseau de neurones convolutifs
- La création
- Courant
- courbe
- Cut/Taille
- Coupe
- cycles
- données
- l'analyse des données
- démontré
- décrit
- Conception
- un
- Avec nos Bagues Halo
- détail
- détails
- Détection
- Déterminer
- déterminé
- déviation
- différence
- numériquement
- directement
- distribution
- adn
- down
- chacun
- de manière efficace
- efficacité
- efficace
- ENGINEERING
- assurer
- erreur
- Ether (ETH)
- Pourtant, la
- événements
- exemple
- expliqué
- extrait
- extraction
- Extraits
- filtres
- finalement
- Prénom
- fleur
- suivi
- Abonnement
- formulaire
- Avant
- De
- plein
- plus
- génome
- en verre.
- guide
- très
- HTTPS
- humain
- Hybride
- Identification
- image
- Imagerie
- la mise en oeuvre
- in
- inclus
- Y compris
- incubé
- INCUBATION
- d'information
- instrument
- interactions
- interne
- Ionique
- IT
- KDA
- Libellé
- conduire
- Longueur
- LINK
- lié
- Location
- lune
- click
- maître
- Matériel
- des mesures
- Mica
- pourrait
- minimum
- mixte
- Mixage audio
- mélange
- ML
- Breeze Mobile
- Mode
- MOL
- plusieurs
- nano
- nanopore
- indigène
- Nature
- négatif
- réseau et
- Neural
- Réseau neuronal
- ouvert
- optimaux
- autrement
- appariement
- patientforward
- patients
- performant
- Physique
- image
- Plastique
- Platon
- Intelligence des données Platon
- PlatonDonnées
- portefeuille
- positif
- Positivité
- défaillances
- Préparer
- préparé
- présence
- empêcher
- précédemment
- apprêt
- sonde
- processus
- propriétés
- Protéines
- protocole
- qualité
- raw
- nous joindre
- réaction
- en temps réel
- région
- régions
- de Presse
- restant
- Supprimé
- Rapports
- un article
- Résultats
- inverser
- ARN
- roche
- Salle
- rt
- Courir
- Sécurité
- même
- SRAS-CoV-2
- balayage
- Section
- choisi
- Séquence
- Shorts
- montrer
- montré
- signaux
- importance
- simultané
- unique
- site
- Sites
- petit
- Logiciels
- quelques
- groupe de neurones
- pivoté
- stable
- Étape
- Utilisation d'un
- Standard
- A déclaré
- statistique
- Étapes
- stockée
- Colliers de perles
- Par la suite
- Ces
- Système
- table
- prise
- Target
- objectifs
- modèle
- Dix
- Le
- ainsi
- trois
- fois
- TM
- à
- ensemble
- Total
- suivre
- Ultra
- sous
- unités
- Universel
- validé
- Plus-value
- Valeurs
- vérifié
- version
- via
- virus
- visualisation
- Tension
- W
- Eau
- web
- application Web
- Wells
- qui
- sans
- wu
- zéphyrnet