جوجه کشی
تمام فرآیندهای جوجه کشی در ترمومیکسر C (Eppendorf) به جز انجیر تکمیلی انجام شد. 1 و 9 (انکوباتور Dry Bath FB15103، Fisher Scientific) و برای شکل. 1c و انجیرهای مکمل. 3-6 (QuantStudio5، Applied Biosystems توسط Thermo Fisher Scientific).
خودآرایی ایزوترمال اوریگامیس DNA دوبعدی
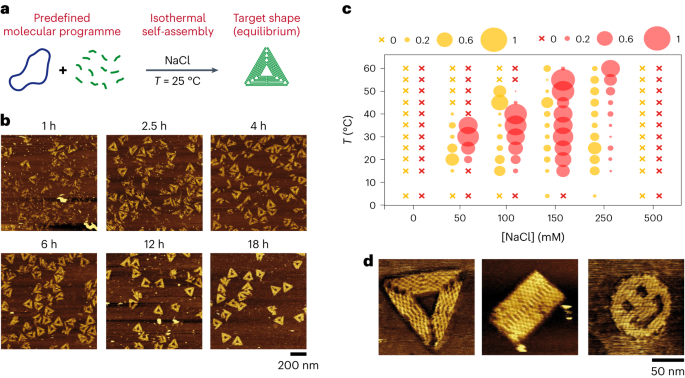
شکل را ببینید. 1 و انجیرهای مکمل. 1-9 و 11-14. ما از کوکتل اصلی بدون هیچ پیش تیمار حرارتی استفاده کردیم و قبل از گرداب کوتاه و افزودن الگوی M13 (1 nM) به محلول و مخلوط کردن ملایم بالا و پایین با پیپت، مستقیماً آن را با بافر مورد نظر مخلوط کردیم. محلول بدون اختلاط بیشتر در دمای ثابت برای مدت زمان مورد نظر در انکوبه گذاشته شد.
بازپخت حرارتی اریگامیس DNA در بافر TANA
به شکل تکمیلی مراجعه کنید. 10. ما الگوی M13 (1 nM) را با مخلوطی از منگنه ها (هر منگنه 40 nM) در TANa همراه با 100mM NaCl مونتاژ کردیم. نمونه به مدت 10 دقیقه در دمای 90 درجه سانتیگراد انکوبه شد و سپس در یک ترموسایکلر peqSTAR 2X (Peqlab) از دمای 70 تا 20 درجه سانتیگراد با سرعت 1- درجه سانتیگراد در هر 10 دقیقه تحت یک رمپ حرارتی قرار گرفت.
تصفیه با رسوب PEG
انجیرهای تکمیلی را ببینید. 13 و 14. اوریگامی DNA به دست آمده توسط مونتاژ همدما در بافر TANa ([NaCl] = 100 mM) در دمای 25 درجه سانتی گراد از رشته های اصلی خود با رسوب PEG خالص شد. این روش از پروتکل معرفی شده در گزارش قبلی الهام گرفته شده است35. اریگامی های DNA سه بار با محلول PEG 8000 و NaCl رقیق شدند تا به ترتیب به غلظت نهایی 4% w/v و mM 500 برسند. پس از مخلوط کردن ملایم، محلول به مدت 15 دقیقه در دمای اتاق انکوبه شد و در 15,000 سانتریفیوژ شد.g به مدت 15 دقیقه مایع رویی برداشته شد و اریگامی ها به حجم اولیه خود در بافر TANA ([NaCl] = 100 mM) معلق شدند. در صورت لزوم، فرآیند برای دومین تصفیه متوالی تکرار شد.
الکتروفورز ژل اوریگامیس خالص شده
به شکل تکمیلی مراجعه کنید. 13. ما 50 میلیلیتر ژل آگارز (نوع I low EEO، Sigma Aldrich) با 1.5 درصد حاوی 4 میکرولیتر GR-Green 10,000× (Excellgen) در بافر TBE 1× آماده کردیم. پس از سرد شدن ژل، در هر چاهک، 18 میکرولیتر از نردبان DNA 100 جفت باز (New England Biolabs) یا 18 میکرولیتر نمونه همراه با 1× محلول SDS رنگ بارگذاری DNA (Thermo Scientific) وارد کردیم. مهاجرت در ولتاژ 100 ولت به مدت 1 ساعت در یک سلول الکتروفورز 7 سانتی متری پر از بافر TBE 1× انجام شد.
مونتاژ گام به گام همدما
به شکل تکمیلی مراجعه کنید. 24. منگنه های مثلث در سه لات مجزا مونتاژ شدند که هر کدام گوشه بالایی، قسمت میانی و لبه مقابل را کدگذاری می کردند. یک لات (40 نانومولار هر منگنه) با M13 (1 نانومولار) در بافر TANA ([NaCl] = 100 mM) مخلوط شد و سیستم در دمای 25 درجه سانتی گراد بدون مخلوط کردن بیشتر انکوبه شد. هر 24 ساعت، حجم لازم برای تصویربرداری AFM را حذف میکنیم، یک لات کدگذاری برای بخش دیگری از مثلث (هر منگنه 40 نانومتر) اضافه میکنیم و اجازه میدهیم سیستم در دمای 25 درجه سانتیگراد در بافر TANA ([NaCl] = 100mM) انکوبه شود. . ما دو روش مختلف مونتاژ گام به گام را انجام دادیم، از گوشه به طرف مقابل و از یک طرف به گوشه مقابل.
آماده سازی ایزوترمال مثلث های اصلاح شده با استرپتاویدین
شکل را ببینید. 2a و انجیرهای مکمل. 15 و 16. در همان لوله، 1 نانومولار M13، 40 نانومولار از هر یک از مواد اصلی شامل موارد بیوتینیله، و 2 میکرومولار استرپتاویدین را در بافر TANa همراه با 100 میلیمولار نمک طعام مخلوط کردیم. نمونه در دمای 25 درجه سانتیگراد بدون مخلوط کردن بیشتر به مدت 24 ساعت انکوبه شد.
آماده سازی ایزوترمال مستطیل های SST R4
شکل را ببینید. 2b. ما تمام رشته های مستطیل R4 را در بافر تا غلظت نهایی 100 نانومولار در هر رشته در TANa همراه با 100 میلی مولار NaCl مخلوط کردیم. نمونه در دمای 25 درجه سانتیگراد بدون مخلوط کردن بیشتر به مدت 24 ساعت انکوبه شد.
الکتروفورز ژل مستطیل های SST R4
شکل را ببینید. 2b. یک ژل آگارز 1.5 درصد (نوع I کم EEO، Sigma Aldrich) در بافر 0.5× TBE همراه با 11 میلی مولار MgCl تهیه شد.2 و رنگ آمیزی DNA سبز GB. الکتروفورز ژل در یک حمام آب یخ به مدت 2 ساعت با ولتاژ 100 اینچ با استفاده از یک نردبان DNA 1 کیلوبایتی انجام شد. برای خالص سازی، نوار هدف ژل را به قطعات کوچک برش داده و در یک لوله با ستون چرخشی قرار داده و ستون را در دمای 5,000 سانتریفیوژ کرد.g به مدت 10 دقیقه برای تصویربرداری AFM، نمونه شسته شده مستقیماً در یک صفحه میکا به مدت 10 دقیقه در داخل محفظه کنترل شده با جو جذب شد. سپس نمونه با 1 میلی لیتر در 0.5 × TBE + 11 mM MgCl شستشو داده شد.2 و با استفاده از AFM در 0.5× TBE + 11 mM MgCl مشاهده شد2.
آماده سازی ایزوترمال نانوشبکه های DNA
شکل را ببینید. 2c و شکل تکمیلی 17. ما نه الیگونوکلئوتید (1 میکرومولار از هر نوکلئوتید) را در بافر TANA که با 100 میلیمولار یا 150 میلیمولار NaCl تکمیل شده بود، مخلوط کردیم. نمونه در دمای 25 درجه سانتیگراد بدون مخلوط کردن بیشتر به مدت 24 ساعت انکوبه شد.
بازپخت حرارتی اریگامی سه بعدی
شکل را ببینید. 3a و شکل تکمیلی 18. داربست (7,560 nt M13 برای Tb، 8,064 nt M13 برای T1) و مخلوط منگنه (10× بیش از حد در هر منگنه) در بافر حاوی 5 میلیمولار Tris-HCl، pH 8.0، 1 میلیمولار MgCl و 18 میلیمولار MgCl EDTA مخلوط شدند.2. این مخلوط به مدت 65 دقیقه در دمای 15 درجه سانتیگراد حرارت داده شد تا تمام رشته های DNA را دناتوره کند، قبل از اینکه به آرامی در یک گرادیان از 60 درجه سانتیگراد تا 40 درجه سانتیگراد سرد شود، بیش از 41 ساعت برای بازپخت و مونتاژ نانوساختارهای اوریگامی سه بعدی.
TEM با لکه منفی
شکل را ببینید. 3 و شکل تکمیلی 18. برای تعیین خصوصیات TEM، ابتدا نانوساختارهای DNA از آگارز 1 درصد (0.5× TBE، 45 میلیمولار تریسبورات، 1 میلیمولار EDTA، pH 8.3) تکمیلشده با 11 میلیمولار MgCl خالصسازی شدند.2 و 0.5 میلی گرم در میلی لیتر-1 Sybr SAFE. نمونه ها به مدت 3 ساعت روی ژل با بافر جاری 0.5 × TBE، 11 میلی مولار MgCl منتقل شدند.2 در 2.85 اینچ V cm-1 در دمای اتاق. نوارهای مربوط به ساختارهای خودآرایی جدا شده و به ستون چرخشی استخراج ژل DNA (Merck) منتقل شدند و در 5,000 گرم به مدت 5 دقیقه در دمای 4 درجه سانتیگراد سانتریفیوژ شدند. سپس اوریگامیهای خالص شده با جذب روی شبکه پوششدادهشده با کربن درخشنده (کوانتی فویل میکرو تولز) رسوب داده شدند، به مدت 60 ثانیه با محلول اورانیل استات آبی 2% (w/v) رنگآمیزی شدند و سپس با کاغذ صافی بدون خاکستر (VWR) خشک شدند. ). مشاهدات TEM بر روی یک میکروسکوپ تنگستن فلش JEM-1400 که با ولتاژ 120 کیلوولت کار میکرد و مجهز به دوربین Gatan OneView بود، انجام شد.
رقابت ایزوترمال
به شکل تکمیلی مراجعه کنید. 20. قبل از افزودن M40 به مخلوط (غلظت نهایی 100 نانومولار) و اختلاط ملایم، مخلوطهای دو مجموعه اصلی را که برای مثلثها و مستطیلها رمزگذاری میکنند (غلظت نهایی 13 نانومولار برای هر منگنه) با بافر (TANA با 1 میلیمولار NaCl) به طور خلاصه گرداب کردیم. با پیپت نمونه در دمای 25 درجه سانتیگراد بدون اختلاط بیشتر قرار داده شد.
مشاهده AFM از نانوساختارهای DNA در مایع
مشاهدات محیطی AFM با وضوح بالا در بافر نمونه برای همه داده ها و تصاویر AFM نشان داده شده در این مقاله استفاده شد. به جز شکل 4 (به پروتکل اختصاصی زیر مراجعه کنید)، نانوساختارهای DNA بهدستآمده در بافر TANA (اوریگامیس DNA با یا بدون اصلاح پروتئین، مستطیلهای SST R4، نانوشبکههای DNA) بر روی دیسکهای میکای تازه شکافته شده با قطر 10 میلیمتر (گرید Nano-Tec V-1) جذب شدند. مسکوویت، لوازم میکروسکوپی نوآورانه میکرو تا نانو) قبلاً روی یک دیسک فلزی چسبانده شده و با 20 میکرولیتر محلول تتراکلرید اسپرمین (0.1 اینچ در آب میلیکیو) به مدت 10 دقیقه تحت درمان قرار گرفته و ابتدا با آب MilliQANa به وفور شسته شده است. برای جذب نمونه، 15 تا 20 میکرولیتر از نمونه بر روی میکای تازه تیمار شده با اسپرمین گذاشته شد و بهجز آزمایشهای تبدیل همدما به مدت 10 دقیقه جذب شد (شکل XNUMX). 5 و انجیرهای مکمل. 21-23) که در آن زمان به دلیل غلظت کمتر اوریگامیس به 20 دقیقه افزایش یافت. سپس صفحه میکا به آرامی با 200 میکرولیتر از بافر شستشو داده شد تا مواد منگنه و اجسام غیرجذب اضافی حذف شوند. برای جلوگیری از خشک شدن نمونه در حین دستکاری میکا، لایه نازکی از بافر را در بالای نمونه جذب شده باقی گذاشتیم و آن را در دمای اتاق در یک محفظه کنترل شده با اتمسفر (یک ظرف در بسته حاوی یک تکه دستمال مرطوب کیم تک خیس شده با MilliQ) نگهداری کردیم. اب). همین پروتکل با نمونه های حاوی منیزیم انجام شد (شکل تکمیلی XNUMX). 1بافرهای TAEMg و TAMg) با این تفاوت که نمونه ها مستقیماً به مدت 5 دقیقه روی میکای تازه شکافته شده بدون هیچ گونه تیماری جذب شدند. نمونه ها با میکروسکوپ نیروی اتمی Cypher ES (Oxford Instruments) در حالت ضربه زدن با فرکانس رزونانس 17-45 کیلوهرتز در مایع و 0.09 اینچ Nmm مشاهده شدند.-1 نوک ثابت نیرو (BL-AC40TS، Olympus)، با استفاده از حالت تحریک فتوترمال blueDrive. تصاویر خام تحت تفریق پسزمینه چند جملهای، تصحیح سطح صفحه، تراز ردیفی با استفاده از روشهای مختلف و تصحیح اسکار افقی در Gwyddion قرار گرفتند.
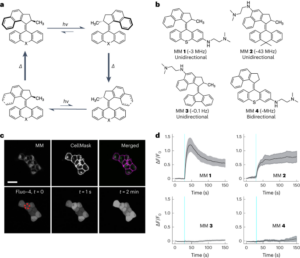
تصویربرداری بلادرنگ از تکامل همدما Λ → Δ روی سطح دولایه لیپیدی
شکل را ببینید. 4، متن تکمیلی 3 و فیلم های تکمیلی 1-4. دولایه های لیپیدی پشتیبانی شده (SLBs) از لیپوزوم های DOPC از طریق روش توصیف شده قبلی ما به دست آمد.37. وزیکول ها از انبار کلروفرم DOPC تهیه شدند. پس از تبخیر کلروفرم تحت جریان گاز نیتروژن، لیپیدها در آب MilliQ دوباره هیدراته شدند تا به غلظت نهایی چربی 2 میلی گرم بر میلی لیتر برسند.-1. سپس مخلوط لیپیدی به مدت 60 دقیقه ورتکس شده و فراصوت شد تا وزیکول های تک لایه کوچک تولید شود. برای جلوگیری از خشک شدن دو لایه، مراحل زیر در یک محفظه کنترل شده با جو انجام شد. SLB ها با قرار دادن 2 میلی لیتر از محلول وزیکول روی دیسک های میکای تازه شکافته شده (که قبلاً روی یک صفحه فلزی مغناطیسی با چسب چسبانده شده بودند) تشکیل شدند، به دنبال آن 2 میکرولیتر TAEMg (تریس استات 1×، [EDTA] = 1 میلیمولار میلیمولار،2] = 12.5 mM). پس از 30 دقیقه جذب، نمونه با 2 میکرولیتر بافر TAEMg برای حذف لیپوزوم های جذب نشده شستشو داده شد و این فرآیند جذب برای بار دوم تکرار شد تا از پوشش بهینه سطح میکا توسط لایه دوتایی اطمینان حاصل شود. در پایان فرآیند جذب، نمونه با 5 میکرولیتر TAENa (تریس استات 1×، [EDTA] = 1 mM، [NaCl] = 100 mM) شستشو شد تا اطمینان حاصل شود که Mg آزاد باقی نخواهد ماند.2+ یون های روی نمونه، که از تا شدن همدما اریگامیس جلوگیری می کند.
اوریگامیس به شکل Λ اصلاح شده با کلسترول (شکل تکمیلی. 19با مخلوط کردن محلولی از 10 نانومولار M13 در بافر TAENa با 20 نانومولار از هر یک از منگنهها و بازپخت با کاهش دما از 70 درجه سانتیگراد به 4 درجه سانتیگراد با سرعت 0.1- درجه سانتیگراد در دقیقه آماده شدند.-1. محلول اریگامی به دست آمده بدون خالص سازی بیشتر مورد استفاده قرار گرفت. سپس، 2 میکرولیتر از محلول اوریگامیس Λ اصلاحشده با کلسترول بر روی SLB از پیش ساخته شده و به دنبال آن 2 میکرولیتر بافر TAENa قرار داده شد. نمونه به مدت 60 دقیقه در دمای اتاق در محفظه کنترل شده اتمسفر انکوبه شد و سپس سطح به طور مستقیم در دمای اتاق تصویربرداری شد.T = 26 °C) توسط AFM در 20 میکرولیتر بافر TAENa بدون شستشوی سطحی. پس از انتخاب یک موقعیت حاوی تعداد کافی ل اوریگامیس جذب شده روی SLB، 8 میکرولیتر از منگنه های سمت A در TAENa بدون جابجایی یا حذف آن از مرحله AFM به نمونه اضافه شد. سپس محلول آویزان نمونه به آرامی با حرکت آهسته پروب AFM به بالا و پایین چندین بار مخلوط شد تا انتشار روی منگنه های سمت A به سمت سطح میکا تسریع شود. سپس همان موقعیت به طور متوسط هر 3 دقیقه به مدت 223 دقیقه اسکن شد. وضوح تصویر 512 × 512 از بود t = 0 به t = 41 min و سپس به 640 × 640 تغییر یافت. z مقیاس تصاویر 0 تا 10 نانومتر بود t = 0 به t = 47 دقیقه و 0-7 نانومتر پس از آن. به دلیل تبخیر در طول فرآیند تصویربرداری، بافر مکمل (8 میکرولیتر) در t = 63 دقیقه، t = 144 دقیقه و t = 161 min، و 8 میکرولیتر از منگنه های سمت A نیز پس از اضافه شدن t = 170 دقیقه
تصاویر AFM در TAENa در دمای اتاق با استفاده از میکروسکوپ نیروی اتمی Brücker Fast Scan در حالت ضربه زدن به دست آمد. تمام آزمایشات AFM با استفاده از پروب های Olympus انجام شد. تصاویر به دست آمده توسط این پروتکل در شکل 1 نمایش داده شده است. 4 و فیلم های تکمیلی 1-4.
تبدیل مورفولوژیکی همدما
شکل را ببینید. 5 و انجیرهای مکمل. 21-23. اوریگامیهای مستطیلی DNA، بدون یا با منگنههای کوتاه شده، ابتدا با بازپخت حرارتی تهیه شدند: پس از مونتاژ 1 نانومولار از الگوی M13 با مخلوطی از منگنهها (هر کدام 40 نانومتر) در بافر TANA (با 100 میلیمولار یا 150 میلیمولار NaCl) نمونه به مدت 10 دقیقه در دمای 90 درجه سانتیگراد انکوبه شد و سپس در یک ترموسایکلر peqSTAR 2X (Peqlab) از دمای 70 درجه سانتیگراد تا 20 درجه سانتیگراد با سرعت 0.1- درجه سانتیگراد در هر 2 دقیقه تحت یک رمپ حرارتی قرار گرفت. سپس منگنه های مثلثی مستقیماً با نمونه مخلوط شدند، بدون هیچ گونه خالص سازی (منگنه های مستطیلی در سیستم نگهداری می شوند)، با یک پیپت به غلظت دلخواه (10 یا 100 نانومولار از هر منگنه) با غلظت نهایی 0.25 نانومولار در [M13] و 10 nM در هر منگنه مستطیل. نمونه به دست آمده در دمای 25 درجه سانتی گراد یا 30 درجه سانتی گراد بدون اختلاط بیشتر قرار داده شد.
آمار و تکرارپذیری
به جز آزمایش نشان داده شده در شکل. 4، که به دلیل پیچیدگی راه اندازی فقط یک بار انجام شد، تمام تحقیقات دیگر چندین بار تکرار شدند تا از تکرارپذیری اطمینان حاصل شود. تمام آنالیزهای تصویر AFM روی تعداد زیادی انجام شد n اریگامی های منفرد گرفته شده از تصاویر مختلف، در موقعیت های مختلف نمونه های به دست آمده در شرایط یکسان. همه اوریگامی های شناسایی شده مورد تجزیه و تحلیل قرار گرفتند. هیچ اوریگامی، به درستی تا شده یا نه، از این تجزیه و تحلیل حذف نشد. شماره n اشیاء تجزیه و تحلیل شده در هر شرایط نشان داده شده در شکل های مختلف در جداول تکمیلی نمایش داده شده است 1-5. هیچ روش آماری برای از پیش تعیین حجم نمونه استفاده نشد. هیچ داده ای از تجزیه و تحلیل حذف نشد. آزمایش ها تصادفی نبودند. محققین در طول آزمایشها و ارزیابی پیامدها نسبت به تخصیص کور نبودند.
- محتوای مبتنی بر SEO و توزیع روابط عمومی. امروز تقویت شوید.
- PlatoData.Network Vertical Generative Ai. به خودت قدرت بده دسترسی به اینجا.
- PlatoAiStream. هوش وب 3 دانش تقویت شده دسترسی به اینجا.
- PlatoESG. خودرو / خودروهای الکتریکی، کربن ، CleanTech، انرژی، محیط، خورشیدی، مدیریت پسماند دسترسی به اینجا.
- BlockOffsets. نوسازی مالکیت افست زیست محیطی. دسترسی به اینجا.
- منبع: https://www.nature.com/articles/s41565-023-01468-2
- :است
- :نه
- :جایی که
- $UP
- 1
- 10
- 100
- 11
- 12
- ٪۱۰۰
- 2%
- 20
- 200
- 2014
- 2015
- 24
- 25
- 26
- 2D
- 30
- 3d
- 40
- 50
- 500
- 60
- 65
- 7
- 70
- 8
- 90
- a
- شتاب دادن
- اضافه
- اضافه کردن
- اضافه
- اضافی
- پس از
- بعد از آن
- معرفی
- تخصیص
- مقدار
- an
- لنگر
- و
- هر
- اعمال می شود
- هستند
- مقاله
- مونتاژ
- مجلس
- ارزیابی
- At
- میانگین
- زمینه
- باند
- BE
- زیرا
- بودن
- در زیر
- BP
- بطور خلاصه
- بافر
- by
- دوربین
- انجام
- سلول
- اتاق
- تغییر
- کلیک
- کوکتل
- برنامه نویسی
- ستون
- پیچیدگی
- غلظت
- شرط
- شرایط
- متوالی
- ثابت
- ظرف
- گوشه
- متناظر
- پوشش
- برش
- رمز
- داده ها
- متراکم
- سپرده
- شرح داده شده
- مطلوب
- شناسایی شده
- مختلف
- انتشار
- مستقیما
- نمایش داده
- DNA
- انجام شده
- پایین
- خشک
- دو
- در طی
- e
- هر
- ed
- لبه
- پایان
- انگلستان
- اطمینان حاصل شود
- مجهز بودن
- اتر (ETH)
- هر
- تکامل
- جز
- مازاد
- محروم
- تجربه
- آزمایش
- FAST
- انجیر
- شکل
- آمار و ارقام
- پر شده
- فیلتر
- نهایی
- نام خانوادگی
- ثابت
- فلاش
- به دنبال
- پیروی
- برای
- استحکام
- تشکیل
- رایگان
- فرکانس
- از جانب
- بیشتر
- بعلاوه
- GAS
- مهربان
- درجه
- سبز
- توری
- بود
- کیفیت بالا
- افقی
- HTTPS
- i
- if
- تصویر
- تصاویر
- تصویربرداری
- in
- از جمله
- افزایش
- جوجه کشی شده است
- جوجه کشی
- ماشین جوجه کشی
- فرد
- اول
- ابتکاری
- داخل
- الهام بخش
- ابزار
- حد واسط
- به
- معرفی
- تحقیقات
- محققان
- IT
- نگه داشته شد
- نردبان
- بزرگ
- لایه
- ترک کرد
- ارتباط دادن
- مایع
- بارگیری
- خیلی
- کم
- کاهش
- دست کاری
- مارتین
- ماده
- مرک
- فلز
- روش
- روش
- میکا
- میکرو
- میکروسکپ
- میکروسکوپ
- مهاجرت
- مهاجرت
- دقیقه
- مخلوط
- مخلوط
- خلط
- مخلوط
- ML
- حالت
- فیلم ها
- متحرک
- نانو
- فناوری نانو
- طبیعت
- لازم
- جدید
- نه
- عدد
- اشیاء
- مشاهده
- به دست آمده
- of
- OLYMPUS
- on
- یک بار
- ONE
- آنهایی که
- فقط
- مقابل
- بهینه
- or
- دیگر
- ما
- خارج
- نتیجه
- روی
- اکسفورد
- مقاله
- بخش
- میخکوب کردن محکم کردن
- برای
- انجام
- قطعه
- قطعات
- افلاطون
- هوش داده افلاطون
- PlatoData
- موقعیت
- موقعیت
- تهیه
- آماده شده
- جلوگیری از
- قبلی
- قبلا
- قبلا
- کاوشگر
- روند
- فرآیندهای
- تولید کردن
- به درستی
- پروتئين
- پروتکل
- قرار دادن
- رمپ
- تصادفی
- نرخ
- خام
- رسیدن به
- کاهش
- باقی مانده
- برداشتن
- حذف شده
- از بین بردن
- مکرر
- تکرار شده
- وضوح
- تشدید
- به ترتیب
- نتیجه
- اتاق
- ROW
- در حال اجرا
- s
- امن
- همان
- مقیاس پذیر
- مقیاس
- اسکن
- علمی
- دوم
- دیدن
- انتخاب
- جداگانه
- مجموعه
- چند
- کوتاه
- نشان داده شده
- طرف
- سیگما
- اندازه
- به آرامی
- کوچک
- راه حل
- مزایا
- خاص
- چرخش
- صحنه
- آماری
- مراحل
- موجودی
- رشته ها
- جریان
- کافی
- پشتیبانی
- سطح
- سیستم
- T
- T1
- صورت گرفته
- ضربه زدن
- هدف
- قالب
- که
- La
- شان
- سپس
- آنجا.
- حرارتی
- اینها
- این
- سه
- زمان
- بار
- نوک
- به
- ابزار
- بالا
- طرف
- منتقل
- دگرگونی
- درمان
- رفتار
- دو
- نوع
- زیر
- استفاده
- با استفاده از
- مختلف
- از طريق
- ولتاژ
- حجم
- بود
- آب
- راه
- we
- خوب
- بود
- که
- اراده
- پاک کردن
- با
- بدون
- کارگر
- خواهد بود
- زفیرنت