Todos los experimentos y procedimientos fueron aprobados por juntas y comités reguladores locales y debían cumplir con los protocolos del estudio. Todos los procedimientos con ratones se realizaron en Caltech, aprobados por el Comité Institucional de Cuidado y Uso de Animales de Caltech (IACUC; protocolo 1738). Los procedimientos para titíes (protocolo TGC-03) y macacos adultos (protocolo LN-14) se llevaron a cabo en el NIH y fueron aprobados por el NIH IACUC. Los procedimientos de tití también se completaron en la Universidad de California en San Diego (UCSD) (protocolo S09147) y cumplieron y aprobaron la UCSD IACUC. Los procedimientos con macacos infantiles se llevaron a cabo en el Centro Nacional de Investigación de Primates de California en la Universidad de California Davis y fueron aprobados por su IACUC local (protocolo 22525). Los procedimientos del mono verde se llevaron a cabo en Virscio y fueron aprobados por su IACUC local.
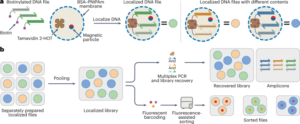
Generación de bibliotecas de ADN de AAV
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5jyl8jy89g2w/v1). Inicialmente generamos diversidad a nivel de ADN, que luego usamos para producir material de transfección para producir la biblioteca de cápside de AAV, como se describió anteriormente en detalle.16. Para la biblioteca de primera ronda, introdujimos esta diversidad genética utilizando cebadores que contienen nucleótidos degenerados insertados entre los aminoácidos 588 y 589 (refs. 12,13,16) (numeración VP1; Fig. complementaria. 1a). Utilizamos un cebador inverso que contiene 7 nucleótidos degenerados ([NNK] × 7) para generar aleatoriamente fragmentos de reacción en cadena de la polimerasa (PCR) que contienen secuencias únicas de 7 unidades insertadas en el tapa gene. Para la biblioteca de ADN de la segunda ronda, utilizamos un grupo de oligos sintéticos (Twist Bioscience) como cebador inverso, que codifica solo las variantes seleccionadas para una mayor detección (en total, 66,628 oligos de ADN; 33,314 variantes recuperadas después de las selecciones de la primera ronda más un codón modificado). réplica de cada uno). Todos los cebadores inversos contenían un saliente 20' de 5 pb complementario al tapa secuencia cerca de la secuencia de la enzima de restricción AgeI y se emparejaron con un cebador directo que contenía un saliente 20' de 5 pb cerca de la secuencia de la enzima de restricción XbaI. Luego insertamos los fragmentos de PCR que contienen la región diversificada en el plásmido rAAV-ΔCAP-in-cis-Lox mediante el ensamblaje de Gibson para generar la biblioteca de ADN de AAV resultante, es decir, rAAV-CAP-in-cis-Lox, utilizando el ensamblaje de ADN NEBuilder HiFi. Mezcla maestra (New England Biolabs, E2621).
Producción de biblioteca de cápside de AAV
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5jyl8jyz9g2w/v1). Generamos bibliotecas de cápsides de AAV de acuerdo con protocolos publicados previamente.16,70. Brevemente, transfectamos células HEK293T (ATCC, cat # CRL-3216; RRID: CVCL_0063) en placas de cultivo de tejidos de 150 mm utilizando polietilenimina lineal de grado de transfección (PEI; Polysciences). En cada placa, transfectamos cuatro plásmidos: (1) la biblioteca de ADN de AAV rAAV-Cap-in-cis-Lox ensamblada, que está flanqueada por repeticiones terminales invertidas necesarias para la encapsidación de AAV; (2) AAV2/9 REP-AAP-ΔCAP, que codifica las proteínas suplementarias REP y AAP necesarias para la producción de AAV con el extremo C del tapa gen escindido para evitar la recombinación con la biblioteca de ADN de AAV y la posterior producción de AAV con capacidad de replicación; (3) pHelper, que codifica las proteínas adenovirales necesarias para la producción de AAV; y (4) pUC18 (Addgene ID: 50004; RRID: Addgene_50004), que no contiene ningún vector de expresión de mamíferos pero se usa como ADN de relleno para lograr la proporción adecuada de nitrógeno a fosfato para una transfección óptima de PEI. Durante la preparación de la mezcla PEI-ADN, agregamos 10 ng de nuestra biblioteca de ADN de AAV (rAAV-Cap-in-cis-Lox) por cada placa de 150 mm y combinamos AAV2/9 REP-AAP-ΔCAP, pUC18 y pHelper en un Relación 1:1:2 (40 µg de ADN total por placa de 150 mm). A las 60 h después de la transfección, purificamos la biblioteca de la cápside de AAV tanto del sedimento celular como del medio utilizando precipitación con polietilenglicol y ultracentrifugación en gradiente de iodixanol. Luego, mediante PCR cuantitativa, determinamos el título de las bibliotecas de cápside de AAV amplificando genomas virales resistentes a DNasaI en relación con un estándar de genoma linealizado de acuerdo con protocolos establecidos.70.
Experimentos con titíes
Selecciones de la biblioteca de cápside
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Todos los titíes (C. jacchus) los procedimientos se realizaron en el Instituto Nacional de Salud Mental (NIMH) y fueron aprobados por el IACUC local. Los titíes nacieron y se criaron en colonias NIMH y se alojaron en grupos familiares en condiciones estándar de 27 °C y 50% de humedad. Fueron alimentados ad libitum y recibieron enriquecimiento como parte del programa de enriquecimiento de primates para NHP en los NIH. Para todos los titíes utilizados en este estudio, no hubo anticuerpos neutralizantes detectables en una dilución de suero de 1:5 antes de las infusiones intravenosas (ensayado por Penn Vector Core, Universidad de Pensilvania). Luego fueron alojados individualmente durante varios días y aclimatados a una nueva habitación antes de las inyecciones. Se utilizaron cuatro hombres adultos para la selección de la biblioteca, dos para cada una de las bibliotecas de primera y segunda ronda. El día anterior a la infusión se retiró la comida de los animales. Los animales fueron anestesiados con isoflurano en oxígeno, la piel sobre la vena femoral se afeitó y se desinfectó con un exfoliante de isopropanol y 2 × 1012 Se infundieron vg de la biblioteca de la cápside de AAV durante varios minutos. Se retiró la anestesia y los animales fueron monitoreados hasta que se volvieron activos, momento en el que fueron devueltos a sus jaulas. La actividad y el comportamiento fueron monitoreados de cerca durante los siguientes 3 días, con observaciones diarias a partir de entonces.
4 semanas después de la inyección, los titíes fueron sacrificados (Eutanasia, VetOne) y perfundidos con solución salina tamponada con fosfato (PBS) 1x. Después de la biblioteca de la primera ronda, el cerebro se cortó en cuatro bloques coronales, se congeló instantáneamente en 2-metilbutano (Sigma-Aldrich, M32631), se enfrió con hielo seco y se almacenó a -80 °C para un almacenamiento a largo plazo. Después de la biblioteca de la segunda ronda, el cerebro se cortó en seis bloques coronales y, junto con secciones de la médula espinal y el hígado, se congeló instantáneamente y se almacenó a -80 °C para un almacenamiento a largo plazo.
Caracterización individual de AAV en titíes.
Los detalles de los procedimientos en esta sección se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 y https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Dos titíes comunes adultos (C. jacchus) se utilizaron para este experimento: Conan (hombre, 2.8 años, 0.386 kg) y Sandy (mujer, 5.8 años, 0.468 kg) (Tabla complementaria 3 proporciona más detalles). Fueron alojados en condiciones estándar de 27 °C y 50% de humedad, con acceso ad libitum a comida y agua. Todos los animales fueron alojados en grupos y los experimentos se realizaron en el Laboratorio de Comportamiento y Sistemas Corticales de la UCSD. Todos los experimentos fueron aprobados por la UCSD IACUC. El día anterior a la infusión se retiró la comida de los animales.
Los animales fueron anestesiados con ketamina (Ketaset, Zoetis 043-304, 20 mg kg-1), la piel sobre la vena safena se afeitó y desinfectó con un exfoliante de isopropanol y 2 × 1013 kg-1 de AAV se infundió durante 5 min. Los animales fueron monitoreados hasta que se volvieron activos, momento en el que fueron devueltos a sus jaulas. La actividad y el comportamiento fueron monitoreados de cerca durante los siguientes 3 días, con observaciones diarias a partir de entonces. Se tomaron muestras de sangre los días 1, 7, 14, 21 y 31 para medir la concentración viral en plasma.
31 días después de la inyección, los titíes fueron anestesiados con ketamina como se describió anteriormente y luego sometidos a eutanasia (Euthasol, Virbac 200-071, 1 ml kg-1) y perfundido con 1× PBS. Los cerebros y órganos se cortaron por la mitad y la mitad se congeló instantáneamente en 2-metilbutano (Sigma-Aldrich, M32631), se enfrió con hielo seco y se almacenó a -80 °C. La otra mitad se fijó en paraformaldehído (PFA) al 4 % (Thermo Scientific, J19943-K2) durante la noche y luego se almacenó a 4 °C en azida de PBS (Sigma-Aldrich, S2002-100G, 0.025 %). Luego, las muestras se enviaron al Instituto de Tecnología de California (Caltech) para su análisis. Para la tinción de GLUT1, incubamos rodajas con anti-GLUT1 de conejo (1:200; Millipore-Sigma, cat # 07-1401; RRID: AB_1587074), realizamos de tres a cinco lavados con PBS, incubamos con IgG anti-conejo de burro (1: 200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) y se lavaron de tres a cinco veces antes del montaje. Diluimos todos los anticuerpos y realizamos todas las incubaciones usando PBS suplementado con Triton X-0.1 al 100% (Sigma-Aldrich, T8787) y suero de burro normal al 10% (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) durante la noche a temperatura ambiente con agitación.
Extracción de ADN de biblioteca viral y preparación de muestras NGS.
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Anteriormente informamos que el ADN de la biblioteca viral y el ARN endógeno del huésped se pueden aislar usando TRIzol precipitando el ácido nucleico de la fase acuosa.12,16. Por lo tanto, para extraer el ADN de la biblioteca viral del tejido de tití, homogeneizamos 100 mg de médula espinal, hígado y cada bloque coronal del cerebro en TRIzol (Life Technologies, 15596) usando un BeadBug (Benchmark Scientific, D1036) y aislamos ácidos nucleicos del fase acuosa según el protocolo recomendado por el fabricante. Tratamos el precipitado reconstituido con RNasa (Invitrogen, AM2288) y lo digerimos con SmaI para mejorar la recuperación del ADN viral aguas abajo mediante PCR. Después de la digestión, purificamos con un kit Zymo DNA Clean and Concentrator (D4033) de acuerdo con el protocolo recomendado por el fabricante y almacenamos el ADN viral purificado a -20 °C.
Para agregar adaptadores de Illumina que flanquean la región diversificada, primero amplificamos por PCR la región que contiene nuestra inserción de 7 unidades utilizando el 50% del ADN viral extraído total como plantilla (25 ciclos). Después de la purificación del ADN de Zymo, diluimos las muestras a 1:100 y las amplificamos aún más alrededor de la región variable con diez ciclos de PCR, agregando regiones de unión para la siguiente reacción de PCR. Finalmente, agregamos adaptadores de celda de flujo de Illumina e índices únicos utilizando NEBNext Dual Index Primers (New England Biolabs, E7600) mediante diez ciclos más de PCR. Luego purificamos en gel los productos finales de la PCR utilizando un gel de agarosa de bajo punto de fusión al 2% (Thermo Fisher Scientific, 16520050) y recuperamos la banda de 210 pb.
Solo para la biblioteca de segunda ronda, también aislamos el ADNss de la biblioteca de AAV encapsidado para NGS para calcular las puntuaciones de enriquecimiento de la biblioteca, una métrica cuantitativa que usamos para normalizar las diferencias en el título de las diversas variantes en nuestra biblioteca (ver ref. 16 y la sección 'Alineación de lectura NGS, análisis y generación de gráficos de red'). Para aislar los genomas virales encapsidados, tratamos la biblioteca de la cápside de AAV con ADNasaI y digerimos las cápsides usando proteinasa K. Luego purificamos el ADN ss usando fenol-cloroformo, amplificamos los transgenes virales mediante dos pasos de amplificación por PCR para agregar adaptadores e índices para Illumina NGS y purificamos. mediante electroforesis en gel. El ADN de esta biblioteca viral, junto con el ADN viral extraído del tejido, se envió para una secuenciación profunda utilizando un sistema Illumina HiSeq 2500 (Laboratorio de Genética y Genómica de Millard y Muriel Jacobs, Caltech).
NGS lee alineación, análisis y generación de gráficos de red.
Los archivos FASTQ sin procesar de ejecuciones de NGS se procesaron con scripts personalizados (https://github.com/GradinaruLab/protfarm y https://github.com/GradinaruLab/mCREATE)16. Para la biblioteca de primera ronda, el proceso para procesar estos conjuntos de datos implicó filtrar para eliminar lecturas de baja calidad, utilizar una puntuación de calidad para cada secuencia y eliminar el sesgo de mutaciones inducidas por PCR o alto contenido de GC. Luego, el conjunto de datos filtrado se alineó mediante un algoritmo de coincidencia de cadenas perfecta y se recortó para mejorar la calidad de la alineación. Luego mostramos los recuentos de lectura absolutos para cada variante durante la secuenciación dentro de cada tejido, y las 33,314 variantes que se encontraron en el cerebro se eligieron para las selecciones de segunda ronda.
Después de las selecciones de la segunda ronda, realizamos el mismo análisis para mostrar el recuento de lectura absoluta de variantes de la biblioteca de virus inyectados y de cada variante dentro de cada tejido. Además, calculamos el enriquecimiento de la biblioteca.16 para cada variante dentro de cada tejido:
$${overline{{rm{RC}}}}_{x,{rm{inyectado}},{rm{biblioteca}}}=,frac{{{rm{RC}}}_{x,{rm{ inyectado}},{rm{biblioteca}}}}{mathop{suma}nolimits_{i=1}^{{N}_{{rm{inyectado}},{rm{biblioteca}}}}{{rm{RC }}}_{i,{rm{inyectado},{biblioteca}}}},$$
(1)
$${overline{{rm{RC}}}}_{x,{rm{tejido}}}=,frac{{{rm{RC}}}_{x,{rm{virus}}}}{mathop {suma }nolimits_{i=1}^{{N}_{{rm{tejido}}}}{{rm{RC}}}_{i,{rm{tejido}}}},$$
(2)
$${rm{Biblioteca},{enriquecimiento}}=,{log }_{10}left(frac{{overline{{rm{RC}}}}_{x,rm{{inyectado},{biblioteca}} }}{{overline{{rm{RC}}}}_{x,{rm{tejido}}}}derecha),$$
(3)
tal que para una muestra dada y (por ejemplo, la biblioteca de virus inyectada o una muestra de tejido), RCx,y es el recuento absoluto de lecturas de la variante x, Ny es el número total de variantes recuperadas y ({sobrelínea{{rm{RC}}}}_{x,{y}}) es el recuento de lecturas normalizado.
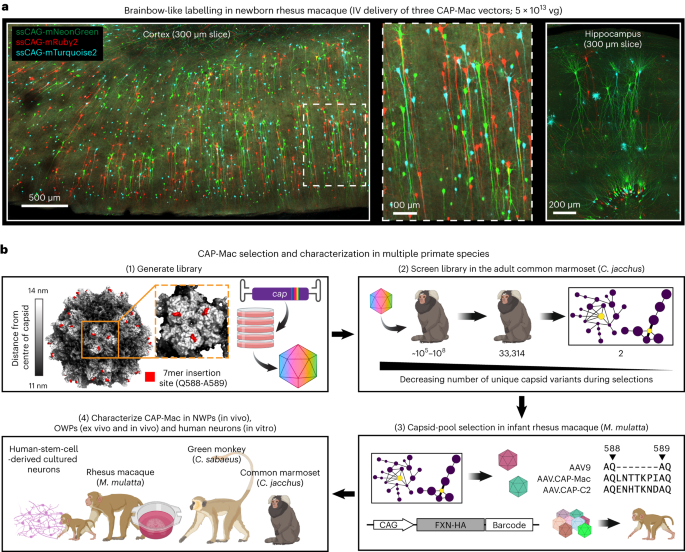
Para construir el gráfico de agrupamiento de secuencias CAP-Mac, filtramos los datos de NGS de la segunda ronda según los siguientes criterios: (1) recuento de lecturas ≥100 en la muestra de biblioteca inyectada (24,186/33,314 variantes), (2) enriquecimiento de biblioteca ≥0.7 puntuación en más de dos muestras de cerebro (415 variantes) y (3) al menos dos muestras de cerebro más con un enriquecimiento de biblioteca ≥0.7 que las muestras de cerebro con un enriquecimiento de biblioteca de menos de -0.7 (323 variantes). Para construir el gráfico de secuencia CAP-C2, filtramos los datos de NGS de la segunda ronda según los siguientes criterios: (1) recuento de lecturas ≥100 en la muestra de la biblioteca inyectada y (2) ambas réplicas de codones presentes en al menos dos muestras de cerebro con Enriquecimiento de biblioteca ≥0.7 (95 variantes). Luego, estas variantes se procesaron de forma independiente para determinar distancias de Hamming inversas por pares (https://github.com/GradinaruLab/mCREATE) y agrupados usando Cytoscape (v. 3.9.0; RRID: SCR_003032) como se describió anteriormente en detalle16. Las redes presentadas muestran variantes de la cápside (nodos) conectadas por bordes si la distancia de Hamming inversa por pares es ≥3.
Clonación de variantes individuales de la cápside de AAV
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.n2bvj87ebgk5/v1). Para la caracterización de una sola variante, clonamos nuevos plásmidos variantes digiriendo una versión modificada de la columna vertebral pUCmini-iCAP-PHP.eB (Addgene ID: 103005; RRID: Addgene_103005) utilizando MscI y AgeI. Diseñamos un cebador de 100 pb que contenía la inserción deseada de 21 pb para cada variante de la cápsida y las regiones complementarias a la plantilla de AAV9 con ~20 pb superpuestos al esqueleto digerido. Luego ensamblamos el plásmido variante usando NEBuilder HiFi DNA Assembly Master Mix, combinando 5 μl de cebador 200 nM con 30 ng de columna principal digerida en la mezcla de reacción. El plásmido de la cápside utilizado para producir AAV.CAP-Mac está disponible en Addgene (Addgene ID: 200658; RRID: Addgene_200658).
Producción y purificación individuales de AAV.
Los detalles de este procedimiento se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.14egn2dqzg5d/v1). Para producir variantes para pruebas conjuntas, seguimos nuestro protocolo publicado anteriormente.70 utilizando placas de cultivo de tejidos de 150 mm. Para la caracterización individual de AAV.CAP-Mac y AAV9 in vivo e in vitro, adoptamos nuestro protocolo publicado para utilizar CellSTACK de diez capas (Corning, 3320) para producir virus de manera eficiente con un título alto para dosificar macacos rhesus y monos verdes. Específicamente, pasamos veinte placas de 150 mm con aproximadamente un 70% de confluencia a un CellSTACK de diez capas 24 h antes de la transfección. El día de la transfección, preparamos la mezcla de transfección PEI-ADN para cuarenta placas de 150 mm, combinamos la mezcla de transfección con medios y realizamos un cambio completo de medios para CellSTACK. Recogimos y cambiamos los medios a las 72 h después de la transfección de manera similar a la producción en platos de 150 mm. A las 120 h después de la transfección, agregamos ácido etilendiaminotetraacético (Invitrogen, 15575020) a una concentración final de 10 mM y lo incubamos a 37 °C durante 20 minutos, agitando y golpeando ocasionalmente los lados del CellSTACK para separar las células. Luego retiramos el medio y la mezcla celular y procedimos con el protocolo de purificación de AAV.70. Es de destacar que durante el paso de intercambio de tampón después de la ultracentifugación, utilizamos concentradores de proteínas centrífugos con membranas de polietersulfona (Thermo Scientific, 88533) en lugar de dispositivos de filtración Amicon y utilizamos PBS de Dulbecco suplementado con Pluronic F-0.001 al 68% (Gibco, 24040032).
Experimentos con roedores
Todos los procedimientos con roedores se realizaron en Caltech y fueron aprobados por la IACUC local. Compramos ratones C57BL/6J (cepa #: 000664; RRID: IMSR_JAX:000664), BALB/cJ (cepa #: 000651; RRID: IMSR_JAX:000651) y DBA/2J (cepa #: 000671; RRID: IMSR_JAX:000671) (todos machos, de 6 a 8 semanas de edad) del Laboratorio Jackson. Para la administración intravenosa en ratones, entregamos 5 × 1011 vg de virus a través del seno retroorbitario70,71 utilizando una jeringa de insulina de calibre 31 (BD, 328438). Consulte protocolos.io para obtener más detalles sobre las inyecciones retroorbitales de AAV en ratones (https://doi.org/10.17504/protocols.io.3byl4joy8lo5/v1). Para la administración intracerebroventricular (ICV) en ratones, inyectamos 5.0 × 1010 o 1.5 × 1011 vg hacia el ventrículo lateral. Brevemente, anestesiamos ratones usando isoflurano (5% para inducción, 1-3% para mantenimiento) con 95% de O2/ 5% CO2 (1 litro min.-1) y se fijó la cabeza de los ratones en un marco estereotáxico. Después de afeitar la cabeza y esterilizar la zona con clorohexidina, administramos por vía subcutánea 0.05 ml de 2.5 mg ml.-1 bupivacaína, se hizo una incisión en la línea media y se limpió el cráneo de sangre y tejido conectivo. Después de nivelar la cabeza, se perforaron bilateralmente orificios por encima de los ventrículos laterales (0.60 mm por detrás del bregma y 1.15 mm desde la línea media). Los vectores virales se aspiraron en jeringas NanoFil de 10 µl (World Precision Instruments) utilizando una aguja de microinyección de calibre 33, y la aguja se bajó lentamente hacia el ventrículo lateral (a 1.6 mm de la superficie pial). Se dejó que la aguja reposara en su lugar durante aproximadamente 5 minutos y se inyectaron de 3 a 5 µl de vector viral utilizando una bomba de microjeringa (World Precision Instruments, UMP3) y un controlador de bomba (World Precision Instruments, Mircro3) a una velocidad de 300 nl min.-1. Todos los ratones recibieron intraoperatoriamente 1 mg kg-1 de buprenorfina SR y 5 mg kg-1 de ketoprofeno por vía subcutánea y 30 mg kg-1 de ibuprofeno y 60 mg kg-1 de trimetoprima/sulfametoxazol durante 5 días después de la cirugía. Consulte protocolos.io para obtener más detalles sobre las inyecciones ICV de AAV en ratones (https://doi.org/10.17504/protocols.io.5qpvorm4dv4o/v1). Después de 3 semanas de expresión, todos los ratones fueron perfundidos con PBS y fijados en PFA al 4%. Todos los órganos se extrajeron, se incubaron en PFA al 4.00% durante la noche, se transfirieron a PBS suplementado con azida sódica al 0.01% y se almacenaron a 4 °C para un almacenamiento a largo plazo. Cortamos el cerebro en secciones de 100 μm con un vibratomo (Leica Biosystems, VT1200S), lo montamos en Prolong Diamond Antifade (Invitrogen, P36970) y tomamos imágenes con un microscopio confocal (Zeiss, LSM 880) usando ZEN (edición negra). Consulte protocolos.io para obtener más detalles sobre el manejo de tejidos (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 y https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1).
Experimentos con macacos Rhesus
Los detalles de los procedimientos en esta sección se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1). Los macacos recién nacidos (0.45-1.40 kg) fueron destetados al nacer. Durante el primer mes, a los macacos se les infundieron vectores AAV por vía intravenosa o intratecal. A todos los macacos adultos (de 8 a 17 años; de 4.65 a 11 kg) incluidos en este estudio se les infundió AAV únicamente mediante administración intravenosa. Para las inyecciones intravenosas, los animales se anestesiaron con ketamina (0.10 ml) y se afeitó y desinfectó la piel sobre la vena safena. AAV (entre 2 × 1013 y 1 × 1014 kg-1) se infundió lentamente en la vena safena durante ~1 min en <0.75 ml de PBS. Para las inyecciones de ICM, a los animales se les administró un sedante por vía intramuscular y se afeitó y preparó asépticamente la zona de la piel del cuello. Se avanzó una aguja hacia la cisterna magna para extraer una pequeña cantidad de LCR proporcional a la cantidad de líquido inyectado. Luego, se extrae una jeringa estéril que contiene la preparación estéril del AAV (1.5 × 1012 o 2.5 × 1013 kg-1) proporcional a la cantidad de líquido recogido se unió asépticamente y se inyectó lentamente. Todos los animales fueron monitoreados durante la recuperación de la sedación durante todo el día y luego diariamente para detectar cualquier hallazgo adverso. Todos los monos fueron alojados individualmente a la vista y el oído de sus congéneres. El tejido se recogió entre 4 y 11 semanas después de la inyección. Los animales fueron anestesiados profundamente y recibieron pentobarbital sódico de acuerdo con las pautas para la eutanasia humana de animales en el Centro Nacional de Investigación de Primates de California. Todo el material inyectado en macacos rhesus estaba libre de endotoxinas (<0.1 UE ml-1), y la pureza de la proteína se confirmó mediante electroforesis en gel de dodecilsulfato de sodio-poliacrilamida. Tablas complementarias 4 y 5 enumere la vía de administración, las variantes de AAV, la dosis viral, la carga genética y la duración de la expresión de cada experimento.
Pruebas en piscinas en macacos rhesus
Los detalles de los procedimientos en esta sección se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1, https://doi.org/10.17504/protocols.io.3byl4jo68lo5/v1 y https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Los experimentos en grupos de macacos neonatos (RM-001 a RM-004) se realizaron en el CNPRC de UC Davis y fueron aprobados por la IACUC local. Los experimentos en grupos de macacos adultos (RMN-001 y RMN-002) se realizaron en el NIMH y fueron aprobados por su IACUC local. Los macacos fueron perfundidos con PBS libre de RNasa helado. En el momento de la perfusión, un hemisferio del cerebro se congeló instantáneamente y el otro hemisferio se seccionó en bloques coronales de 4 mm y se fijó posteriormente en PFA al 4% durante 48 h y se transfirió a Caltech para su posterior procesamiento. Para la tinción con HA, incubamos rodajas con anti-HA de conejo (1:200; Cell Signaling Technology, cat n.° 3724; RRID: AB_1549585), realizamos de tres a cinco lavados con PBS, incubamos con IgG anti-conejo de burro (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) y se lavó de tres a cinco veces antes del montaje. Diluimos todos los anticuerpos y realizamos todas las incubaciones usando PBS suplementado con Triton X-0.1 al 100% (Sigma-Aldrich, T8787) y suero de burro normal al 10% (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) durante la noche a temperatura ambiente con agitación.
Para aislar el ADN viral y el ARN completo, se homogeneizaron cortes de 100 mg de cerebro e hígado en TRIzol (Life Technologies, 15596) utilizando un BeadBug (Benchmark Scientific, D1036) y se recuperó el ADN y el ARN totales según el protocolo recomendado por el fabricante. . El ADN recuperado se trató con RNasa, se digirió por restricción con SmaI y se purificó con un kit Zymo DNA Clean and Concentrator (D4033). El ARN recuperado se trató con ADNasa y se generó ADNc a partir del ARNm utilizando SuperScript III (Thermo Fisher Scientific, 18080093) y cebadores oligo(dT) según el protocolo recomendado por el fabricante. Utilizamos PCR para amplificar la región del código de barras utilizando 50 ng de ADN viral o ADNc como plantilla. Después de la purificación del ADN de Zymo, diluimos las muestras a 1:100 y amplificamos aún más la región del código de barras utilizando cebadores para agregar adaptadores para Illumina NGS. Después de la limpieza, estos productos se amplificaron aún más utilizando NEBNext Dual Index Primers para secuenciación de Illumina (New England Biolabs, E7600) durante diez ciclos. Luego purificamos en gel los productos de PCR finales utilizando un gel de agarosa de bajo punto de fusión al 2% (Thermo Fisher Scientific, 16520050). El enriquecimiento de las pruebas del grupo se calculó de manera idéntica al enriquecimiento de la biblioteca, pero se representa en la Fig. 2b, c en una escala lineal.
Caracterización individual de CAP-Mac en macacos rhesus.
Los detalles de los procedimientos en esta sección se pueden encontrar en protocolos.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 y https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Los macacos recién nacidos fueron perfundidos con PBS y PFA al 4%. El cerebro se seccionó en bloques coronales de 4 mm y todo el tejido se fijó posteriormente en PFA al 4% durante 3 días antes del almacenamiento en PBS. El único macaco adulto utilizado para la caracterización individual in vivo (RM-020; 17 años, 11 kg) fue perfundido con PBS libre de RNasa, y un medio hemisferio se congeló instantáneamente y el otro se seccionó en bloques coronales de 4 mm y postfijado en PFA al 4%. Todo el tejido se transfirió a Caltech para su posterior procesamiento. Los cerebros y los hígados se cortaron en rodajas de 100 μm utilizando un vibratomo. Además, se incubaron secciones de cerebro y médula espinal en sacarosa al 30% durante la noche, se incluyeron en compuesto OCT (Scigen, 4586) y se seccionaron en rodajas de 50 μm utilizando un criostato (Leica Biosystems, CM1950). Todos los cortes se montaron con Prolong Diamond Antifade y se tomaron imágenes con un microscopio confocal. Para la tinción con GFP de cortes de médula espinal y cerebro del macaco administrado por vía intratecal, incubamos cortes con anti-GFP de pollo (1:500; Aves Labs, cat # GFP-1020; RRID: AB_10000240), realizamos de tres a cinco lavados con PBS. , se incubaron con IgY anti-pollo de burro (1:200; Jackson ImmunoResearch Lab, cat # 703-605-155; RRID: AB_2340379) y se lavaron de tres a cinco veces antes del montaje. Diluimos todos los anticuerpos y realizamos todas las incubaciones usando PBS suplementado con Triton X-0.1 al 100% (Sigma-Aldrich, T8787) y suero de burro normal al 10% (Jackson ImmunoResearch, 017-000-121) durante la noche a temperatura ambiente con agitación.
Para la reconstrucción morfológica, dividimos los cerebros en secciones de 300 μm y los incubamos en una solución que coincidiera con el índice de refracción.72 durante 72 h antes de montarlo en un portaobjetos sumergido en la solución de adaptación del índice de refracción. Tomamos imágenes usando un microscopio confocal y un objetivo ×25 (LD LCI Plan-Apochroma ×25/0.8 Imm Corr DIC) usando 100% glicerol como líquido de inmersión. Capturamos mosaicos Z pilas (1,024 × 1,024 para cada fotograma utilizando la configuración de captura sugerida) alrededor de las celdas de interés y campos de visión apropiados recortados para el seguimiento. El rastreo se realizó en Imaris (Oxford Instruments; RRID: SCR_007370) utilizando métodos semiautomáticos y automatizados.
Para la cuantificación de neuronas (NeuN) y astrocitos (S100β), las rodajas se tiñeron usando un anticuerpo anti-NeuN (EPR12763) (1:200; Abcam, cat # ab177487; RRID: AB_2532109) o un anticuerpo anti-S100β (1:200; Abcam , cat # ab52642; RRID: AB_882426) durante la noche en PBS suplementado con Triton X-0.1 al 100 % y suero de burro normal al 10 %. Las rodajas se lavaron de tres a cinco veces con PBS y se incubaron durante la noche en anticuerpo IgG anti-conejo conjugado con Alexa Fluor 647 (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) en PBS + 0.1%. Triton X-100 + 10% suero normal de burro. Después de tres a cinco lavados y montaje con Prolong Diamond Antifade, obtuvimos Z apila utilizando un microscopio confocal y un objetivo de ×25. Segmentamos células positivas para NeuN y XFP utilizando scripts personalizados en Python (RRID: SCR_008394) y Cellpose (https://www.cellpose.org/; RRID: SCR_021716)73.
Imágenes de dos fotones ex vivo
Se prepararon cortes de cerebro de tamaños adecuados para la obtención de imágenes con un espesor de 400 µm a partir de cortes más grandes utilizando un vibratomo y se almacenaron en líquido cefalorraquídeo artificial burbujeado con gas carbógeno antes de obtener imágenes de dos fotones, como se describió anteriormente en protocolos publicados.74,75. Para probar las respuestas de GCaMP8, se administró estimulación eléctrica (4 a 5 V, 80 Hz, 0.3 s de duración) con el número indicado de pulsos utilizando un electrodo monopolar extracelular colocado a 100 a 200 µm de distancia de la neurona de la que se tomaron imágenes. La velocidad de fotogramas de las imágenes fue de 30 Hz. Los rastros de regiones de interés segmentadas se trazaron como ΔF/F0 = (F(t) − F0)/F0, Donde F0 se define como el promedio de todos los valores de fluorescencia antes de la estimulación eléctrica. El tiempo de subida se definió como el tiempo necesario para que la fase ascendente de la señal alcance desde el 10% del pico hasta el 90% del pico. La constante de tiempo de caída se obtuvo ajustando las sumas de exponenciales a la fase de caída de la señal. La relación señal-ruido se obtuvo dividiendo la amplitud máxima de la señal por la desviación estándar de la traza de fluorescencia antes de la estimulación eléctrica.
Caracterización en rodaja de macaco rhesus adulto.
Se planificó la eutanasia de rutina de un macaco rhesus adulto (14 años y 1 mes de edad; 10.83 kg) del Centro Nacional de Investigación de Primates de Washington, y el cerebro se recolectó como parte del Programa de Distribución de Tejidos de la instalación. Se seccionó un bloque de la circunvolución temporal superior en cortes de 300 μm y se recuperaron los cortes.74 y cultivado en una interfaz de membrana aire-líquido, como se describió anteriormente76. Aproximadamente 30 minutos después de sembrar las rodajas, administramos 1 a 2 μl de AAV (5 × 1013 ml-1 del paquete AAV9 o AAV.CAP-Mac, ya sea ssCAG-FXN-HA o ssCAG-eGFP). Los experimentos se realizaron por triplicado biológico para cada condición y el medio de cultivo se actualizó cada 48 h hasta la recolección del tejido a los 8 días después de la transducción. El día de la recolección del tejido, se tomaron imágenes de los cortes para confirmar la transducción, los cortes se cortaron por la mitad y cada mitad se congeló instantáneamente en un baño de hielo seco y etanol. Las muestras se almacenaron a -20 °C hasta su posterior procesamiento.
Se procesó cada media rebanada (una para la recuperación de ADN y otra para la recuperación de ARN). El ADN se aisló utilizando el kit Qiagen DNeasy Blood and Tissue (Qiagen, n.º de catálogo 69504) y el ARN se recuperó utilizando TRIzol (Thermo Fisher Scientific, n.º de catálogo 15596026) y el kit PureLink RNA Mini (Thermo Fisher Scientific, n.º de catálogo 12183018A). El ADN se eliminó de la muestra de ARN modificando el primer lavado del kit PureLink RNA Mini de la siguiente manera: lave con 350 µl de tampón de lavado 1, luego agregue 80 µl de ADNasaI libre de ARNasa en tampón RDD (n.º de catálogo de Qiagen 79254) e incube la columna a temperatura ambiente durante 15 min; luego, lavar nuevamente con 350 µl de Wash Buffer 1 antes de continuar con el protocolo. Realizamos la síntesis de ADNc de primera cadena a partir de 400 ng de ARN total en reacciones de 20 µl utilizando un kit de transcripción inversa Promega GoScript (Promega, n.º de catálogo A5000).
Luego evaluamos los genomas de los vectores y las transcripciones virales encontradas en cada muestra mediante PCR cuantitativa en un Roche Lightcycler II. En este caso, se utilizaron 100 ng de ADN en una reacción de amplificación de 20 µl utilizando sondas TaqMan de Thermo Fisher Scientific (sonda EGFP-FAM, ID de ensayo Mr04097229_mr, n.º de catálogo 4331182; sonda de referencia genómica personalizada CN2386-2-VIC, ID de ensayo ARH6DUK, n.º de catálogo). 4448512, diseñado para apuntar tanto M. mulata y Macaca nemestrina).
Experimentos con el mono verde
Todo el mono verde (C. sabaeus) los procedimientos se realizaron en Virscio y fueron aprobados por su IACUC. Todos los monos fueron examinados en busca de anticuerpos neutralizantes y se confirmó que tenían un título <1:5. Aproximadamente a los 7 a 8 meses de edad (1.0 a 1.3 kg), los monos recibieron una dosis por vía intravenosa (Tabla complementaria 6). Se permitió que las formulaciones de dosis se equilibraran aproximadamente a temperatura ambiente durante al menos 10 minutos, pero no más de 60 minutos antes de la dosificación. Los volúmenes de dosis intravenosas se basaron en el peso corporal del día 0. Los animales fueron sedados con ketamina (8.0 mg kg-1) y xilazina (1.6 mg kg-1). El área de inyección se afeitó y se preparó con clorohexidina e isopropanol al 70% y se lavó quirúrgicamente antes de la inserción del catéter intravenoso. La dosificación se realizó con una única infusión intravenosa de AAV (7.5 × 1013 o 7.6 × 1013 kg-1) el día 0 a través de la vena safena administrada mediante un dispositivo de infusión manual a una velocidad objetivo de 1 ml min.-1. El bienestar general se confirmó dos veces al día mediante observación en la jaula comenzando 1 semana antes de la dosificación. A la hora programada para la eutanasia, los monos fueron sedados con ketamina (8-10 mg kg-1 intramuscular) y sacrificados con pentobarbital sódico (100 mg kg-1 IV al efecto). Ante la pérdida del reflejo corneal, se realizó una perfusión transcardíaca (ventrículo izquierdo) con PBS enfriado utilizando una bomba peristáltica ajustada a una velocidad de aproximadamente 100 ml min.-1 hasta que el líquido que escapa saliera claro antes de la recolección del tejido. Se recolectaron cubos de tejido del hemisferio izquierdo del cerebro y de varios otros órganos y se congelaron en la fase de vapor de nitrógeno líquido para su posterior procesamiento para su biodistribución. Se extrajo el hemisferio derecho del cerebro y se cortó en cortes coronales de aproximadamente 4 mm y se fijó intacto con aproximadamente 20 volúmenes de formalina tamponada neutra al 10 % durante aproximadamente 24 h a temperatura ambiente.
El ADN genómico se extrajo del SNC y de tejidos periféricos utilizando el kit de extracción Thermo Fisher MagMax DNA Ultra 2.0 (n.º de catálogo A36570). Se evaluó el rendimiento del ADN mediante cuantificación fluorométrica con el ensayo Qubit dsDNA. Se cargaron aproximadamente 20 ng de ADN en cada reacción de 20 µl y las placas se procesaron en el sistema de detección de PCR en tiempo real BioRad CFX Connect (n.º de catálogo 1855201). La especificidad del ensayo del número de copias virales se validó mediante la detección de un único producto amplificado; sensibilidad, evaluando que el límite inferior de detección sea superior a diez copias por reacción; y linealidad, asegurando la curva estándar R2 fue >0.95. Las reacciones se ensamblaron en FastStart Universal SYBR Green Master (Rox) (catálogo n.° 4913850001). Las secuencias de los cebadores fueron ACGACTTCTTCAAGTCCGCC (adelante) y TCTTGTAGTTGCCGTCGTCC (inversa). El protocolo de PCR utilizó un paso de desnaturalización inicial de 95 °C durante 180 s, seguido de 40 ciclos de 95 °C durante 15 s y 60 °C durante 60 s, con un paso de obtención de imágenes después de cada ciclo de 60 °C. Se generó una curva estándar con plásmido linealizado que contiene la secuencia plantilla de GFP presente en el virus de 1 × 108 a 1 × 1010 copias, diluidas en muestras de ADN de macaco no tratadas y preparadas utilizando un kit idéntico al de las muestras de este estudio para controlar los efectos de la matriz. Las copias de ADN viral se calcularon a partir de la curva estándar utilizando la ecuación para la línea de mejor ajuste. Los valores de multiplicidad de infección se calcularon en función del peso genómico total medido del ADN de la célula huésped por reacción.
Después de la fijación, los tejidos se colocaron en 10% > 20% > 30% de sacarosa durante 24 h cada uno a 4 ° C y luego se incluyeron en compuesto OCT y se almacenaron a -80 ° C hasta la criosección. Los bloques de tejido se llevaron a -20 °C en un criostato antes de seccionarlos en rodajas de 30 μm y se montaron en seco en portaobjetos después de la criosección. Después de seccionar, los portaobjetos se dejaron secar a temperatura ambiente durante la noche. Para ayudar en la cuantificación de neuronas, teñimos secciones con los siguientes anticuerpos y concentraciones: anti-GFP de conejo (1:100; Millipore-Sigma, n.º de cat. AB3080; RRID: AB_91337) y anti-NeuN de ratón (A60) (1:500; Millipore-Sigma, número de catálogo MAB377; RRID: AB_2298772). Para la tinción secundaria de anticuerpos, se utilizaron los siguientes anticuerpos secundarios y concentraciones: burro anti-conejo Alexa Fluor 488 (1:500; Thermo Fisher Scientific, cat # A-21206; RRID: AB_2535792) y burro anti-ratón Alexa Fluor 647 (1 :500; Thermo Fisher Scientific, número de catálogo A-31571; RRID: AB_162542). Todos los anticuerpos se diluyeron con 1x PBS suplementado con Triton X-0.25 (PBST) al 100 % y suero de burro normal al 5.00 %. Las incubaciones de anticuerpos primarios se dejaron durante la noche a temperatura ambiente. Luego se lavaron las secciones con PBST. Las incubaciones de anticuerpos secundarios se realizaron durante 2 h a temperatura ambiente. Las secciones se lavaron tres veces en PBST. Las secciones se incubaron en solución DAPI (1:10,000; Invitrogen, D1306) a temperatura ambiente durante 5 minutos y luego se lavaron. Las secciones se cubrieron con Prolong Diamond Antifade.
Se tiñeron y se tomaron imágenes de tres secciones por animal. Se tomaron imágenes de cada sección por triplicado, y cada región de interés tenía un total de nueve imágenes. Se tomaron imágenes de las regiones de interés del tejido con un Keyence BZ-X800 con los siguientes parámetros de adquisición: GFP (1/500 s), Cy5 (1 s), DAPI (1/12 s), alta resolución Z apilar con un paso de 1.2 µm. Se tomaron imágenes de las siguientes subregiones del cerebro: corteza frontal, parietal, temporal, occipital, cerebelo, caudado, putamen y tálamo (núcleos medial, ventral lateral y ventral posterior). Se realizó un método de recuento de células semiautomático utilizando ImageJ (RRID: SCR_003070) para la cuantificación. Utilizando umbrales y análisis de partículas, cuantificamos células positivas para NeuN y positivas para DAPI. Utilizando el contador de células de ImageJ, contamos manualmente las células positivas para GFP, así como las células doblemente positivas para GFP y NeuN.
experimentos iPSC
Los cultivos neuronales se produjeron diferenciando y madurando células progenitoras neurales derivadas de iPSC con kits de diferenciación y maduración del cerebro anterior Stemdiff (StemCell # 08600 y # 08605, respectivamente), de acuerdo con los protocolos de su fabricante. Las células progenitoras neurales se produjeron mediante diferenciación de la línea iPSC derivada de fibroblastos de prepucio: ACS-1019 (ATCC# DYS-0100; RRID: CVCL_X499), con kits de inducción neuronal Stemdiff SMADi (StemCell l#08581), selección con Stemdiff Neural Rosette Selection. Reactivo (StemCell l#05832) y expansión en Stemdiff Neural Progenitor Media (StemCell l#05833), según los protocolos de su fabricante. Las neuronas maduraron un mínimo de 8 días antes de volver a colocarlas para la transducción.
Los cultivos neuronales maduros, sembrados con 15,000 células por pocillo en placas ópticas de 96 pocillos con paredes negras recubiertas de poliornitina y laminina, se cultivaron durante 4 días adicionales antes de la transducción. Se transdujeron pocillos replicados con virus diluidos en serie en seis órdenes de magnitud en medios de maduración al 90 % y OptiPRO SFM al 10 %. Cuatro días después de la transducción, los cultivos se fijaron con PFA al 4% y se contratiñeron con 1 µg ml-1 Hoechst 33322. La identificación de las células transducidas se determinó mediante la obtención de imágenes de 60 campos por pocillo, utilizando detección de fluorescencia de dos canales (Hoechst, ex386/em440; eGFP, ex485/em521) en una plataforma CellInsight CX5 HCS. Las células individuales se identificaron mediante la detección de Hoechst de sus núcleos y la aplicación de máscaras de anillo de tamaño y contacto restringidos a cada célula. La transducción celular se determinó midiendo la fluorescencia de eGFP por encima de un nivel umbral dentro de una máscara de anillo individual. Para cada población, se representó gráficamente el porcentaje de células transducidas frente a la dosis aplicada. Ajustes de curvas y EC50 los valores se determinaron con el método de regresión agonista versus respuesta (tres parámetros) de Prism GraphPad (RRID: SCR_002798). Para informar las eficiencias de expresión de eGFP por célula, se promediaron las intensidades de fluorescencia puntual de eGFP de cada máscara de anillo en un mínimo de 5,000 células por pocillo. Los ajustes de la curva se obtuvieron utilizando Prism GraphPad Biphasic X como método de regresión de concentración.
Estadística y reproducibilidad
Para las imágenes representativas, se montaron al menos tres cortes separados de cada muestra para obtener imágenes. Dentro de cada región del cerebro de un solo animal, se tomaron al menos tres campos de visión diferentes (campo de visión mínimo después del mosaico, 2.38 mm × 2.38 mm; grosor del corte, 50 µm), lo que equivale a nueve campos de visión separados en tres cortes del cerebro. , para garantizar la coherencia entre las muestras de imágenes.
Resumen de informes
Más información sobre el diseño de la investigación está disponible en el Resumen de informes de la cartera de naturaleza vinculado a este artículo.
- Distribución de relaciones públicas y contenido potenciado por SEO. Consiga amplificado hoy.
- PlatoData.Network Vertical Generativo Ai. Empodérate. Accede Aquí.
- PlatoAiStream. Inteligencia Web3. Conocimiento amplificado. Accede Aquí.
- PlatoESG. Automoción / vehículos eléctricos, Carbón, tecnología limpia, Energía, Ambiente, Solar, Gestión de residuos. Accede Aquí.
- Desplazamientos de bloque. Modernización de la propiedad de compensaciones ambientales. Accede Aquí.
- Fuente: https://www.nature.com/articles/s41565-023-01419-x
- :es
- :dónde
- $ UP
- 000
- 1
- 10
- 100
- 11
- 12
- 13
- 14
- 15%
- 16
- 17
- 180
- 2%
- 20
- 200
- 2011
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 24
- 25
- 27
- 30
- 300
- 31
- 33
- 39
- 40
- 50
- 500
- 60
- 66
- 7
- 70
- 72
- 75
- 8
- 80
- 9
- 95%
- a
- arriba
- Absoluto
- de la máquina
- conformidad
- Conforme
- Lograr
- adquisición
- a través de
- lector activo
- actividad
- Ad
- add
- adicional
- Adicionales
- Adicionalmente
- administrado
- administración
- adoptado
- Adulto
- avanzado
- adverso
- Después
- de nuevo
- edad
- AL
- Alexa
- algoritmo
- alineado
- Todos
- permitido
- a lo largo de
- también
- cantidad
- Amplificación
- Amplificado
- amplificando
- an
- análisis
- Comercial
- y
- animal
- animales
- Anticuerpos
- cualquier
- aplicada
- La aplicación de
- adecuado
- aprobado
- aproximadamente
- Reservada
- en torno a
- artículo
- artificial
- AS
- ensamblada
- Asamblea
- evaluado
- Evaluación
- ayudar
- At
- Confirmación de Viaje
- Hoy Disponibles
- promedio
- lejos
- Columna vertebral
- BANDA
- basado
- BD
- BE
- se convirtió en
- antes
- Comienzo
- "Ser"
- MEJOR
- entre
- parcialidad
- uniéndose
- Negro
- Bloquear
- Bloques
- sangre
- cuerpo
- nacido
- ambas
- BP
- Cerebro
- sesos
- brevemente
- Traído
- buffer
- pero
- by
- jaulas
- calcular
- calculado
- California
- PUEDEN
- capturar
- capturado
- servicios sociales
- CAT
- (SCD por sus siglas en inglés),
- Células
- celulares
- Reubicación
- central
- cadena
- chan
- el cambio
- cambiado
- elegido
- limpiar
- clic
- de cerca
- clustering
- --
- Columna
- combinado
- combinar
- comité
- Algunos
- complementario
- completar
- Completado
- compliance
- cumplir
- Compuesto
- concentración
- condición
- condiciones
- Confirmar
- Confirmado
- Contacto
- conectado
- constante
- construir
- contenida
- contiene
- control
- controlador
- copias
- Core
- coronal
- Para contrarrestar
- criterios
- Cultura
- curva
- personalizado
- Custom-construido
- Corte
- Cycle
- de ciclos
- todos los días
- datos
- conjuntos de datos
- Davis
- día
- Días
- profundo
- se define
- liberado
- entrega
- descrito
- Diseño
- diseñado
- deseado
- detalles
- Detección
- Determinar
- determina
- desviación
- dispositivo
- Dispositivos
- Diamante
- Diego
- diferencias
- una experiencia diferente
- dilución
- Pantalla
- aquí
- distancia
- distinto
- diversificado
- Diversidad
- ADN
- hecho
- dosificar
- dosificación
- el lado de la transmisión
- secas
- duración
- durante
- e
- E & T
- cada una
- Más temprano
- edición
- efecto
- los efectos
- eficiencias
- eficiente
- eficiente.
- ya sea
- elementos
- eliminando
- integrado
- England
- garantizar
- asegurando que
- se establece
- Éter (ETH)
- EU
- evaluado
- Cada
- ejemplo
- Intercambio
- expansión
- experimento
- experimentos
- expresión
- extraerlos
- Extracción
- familia
- Fed
- hembra
- campo
- Terrenos
- Higo
- Figura
- archivos
- filtración
- final
- Finalmente
- Los resultados
- Nombre
- cómodo
- adecuado
- Digital XNUMXk
- fijas
- Flash
- de tus señales
- fluido
- seguido
- siguiendo
- siguiente
- Comida
- por rendimiento
- adelante
- encontrado
- Digital XNUMXk
- FRAME
- Gratis
- Desde
- congelado
- funcional
- promover
- GAS
- calibre
- General
- generar
- generado
- generación de AHSS
- Genética
- genoma,
- genómica
- dado
- gráfica
- mayor
- Verde
- Grupo procesos
- Grupo
- orientaciones
- A Mitad
- Manejo
- Tienen
- es
- cabeza
- Salud
- esta página
- Alta
- de alta resolución
- Agujeros
- homogeneizado
- fortaleza
- HTTPS
- humana
- HIELO
- ID
- idéntico
- Identificación
- no haber aun identificado una solucion para el problema
- if
- ii
- iii
- imágenes
- Proyección de imagen
- inmerso
- inmersión
- mejorar
- in
- incluido
- incubado
- independientemente
- índice
- indicado
- Indices
- INSTRUMENTO individual
- Individualmente
- inducción
- infección
- información
- infundido
- infusión
- inicial
- posiblemente
- Innovadora
- Institucional
- instrumentos
- intereses
- Interfaz
- dentro
- intravenoso
- Introducido
- involucra
- aislado
- Jackson
- kit
- el lab
- laboratorio
- labs
- mayores
- ld
- menos
- izquierda
- menos
- Nivel
- bibliotecas
- Biblioteca
- Vida
- LIMITE LAS
- línea
- LINK
- vinculado
- Líquido
- Lista
- Hígado
- local
- compromiso a largo plazo
- de
- inferior
- bajada
- hecho
- un mejor mantenimiento.
- a mano
- cartografía
- máscara
- Mascarillas
- dominar
- Match
- pareo
- materiales
- Matrix
- medir
- mesurado
- medición
- Medios
- mediano
- mental
- Salud mental
- Método
- métodos
- métrico
- ratones
- Microscopio
- min
- mínimo
- minutos
- mezcla
- mezcla
- ML
- modificado
- monitoreado
- Mes
- meses
- más,
- ARNm
- MSCI
- a saber
- nanotecnología
- Nacional
- Naturaleza
- Cerca
- necesario
- del sistema,
- telecomunicaciones
- Neural
- neuronales
- Neuronas
- Nuevo
- Next
- NIH
- no
- nodos
- normal
- número
- objetivo
- obtenido
- se produjo
- of
- Viejo
- on
- ONE
- , solamente
- óptimo
- optimizado
- or
- en pedidos de venta.
- Otro
- "nuestr
- Más de
- durante la noche
- Oxford
- Oxígeno
- embalaje
- emparejado
- parámetros
- parte
- partícula
- PBS
- PCR
- En pleno
- Penn
- Pennsylvania
- para
- porcentaje
- perfecto
- realizado
- periférico
- fase
- industrial
- Paso
- Colocar
- planificado
- Plasma
- plataforma
- Platón
- Inteligencia de datos de Platón
- PlatónDatos
- más
- alberca
- población
- portafolio
- Precisión
- preparación
- preparado
- presente
- presentó
- evitar
- previamente
- primario
- cartilla
- sonda
- procedimientos
- procedimientos
- procesado
- tratamiento
- producir
- producido
- Producto
- Producción
- Productos
- progenitor
- Programa
- Cobertura
- Proteínas
- Proteínas
- protocolo
- protocolos
- proporciona un
- publicado
- bomba
- comprado
- Python
- calidad
- XNUMX% automáticos
- Qubit
- Conejo
- elevado
- Rate
- proporción
- en comunicarse
- reacción
- reacciones
- Leer
- en tiempo real
- recibido
- recomendado
- recuperación
- región
- regiones
- regresión
- regulador
- relativo
- remove
- Remoto
- réplicas
- reporte
- reportado
- Informes
- representante
- representado
- Requisitos
- la investigación
- respectivamente
- respuesta
- respuestas
- restricción
- resultante
- marcha atrás
- Derecho
- Anillos
- Subir
- creciente
- moléculas de ARN
- robusto
- roche
- Conferencia
- Ruta
- Ejecutar
- corre
- s
- mismo
- San
- San Diego
- arenoso
- Escala
- programada
- SCI
- científico
- Puntuación
- puntuaciones
- proyección
- guiones
- secundario
- Sección
- (secciones)
- ver
- segmentación
- seleccionado
- selección
- selectivo
- Sensibilidad
- expedido
- separado
- Secuencia
- secuenciación
- Serum
- set
- ajustes
- Varios
- transportado
- Mostrar
- Plato Adicional
- Visión
- Signal
- similares
- soltero
- sentarse
- SEIS
- tamaños
- Piel
- Rebanada
- diapositiva
- Slides
- Despacio
- chica
- sodio
- a medida
- Aislamiento de Sonido
- específicamente
- especificidad
- Spot
- montón
- Stacks
- estándar
- paso
- pasos
- STORAGE
- almacenados
- Cordón
- ESTUDIO
- posterior
- adecuado
- superior
- Superficie
- sintético
- te
- sistémico
- Todas las funciones a su disposición
- mesa
- toma
- tocando
- Target
- afectados
- orientación
- Tecnologías
- Tecnología
- plantilla
- diez
- terminal
- Pruebas
- que
- esa
- La
- El Área
- su
- Les
- luego
- Ahí.
- por lo tanto
- Estas
- ellos
- así
- Tres
- umbral
- A través de esta formación, el personal docente y administrativo de escuelas y universidades estará preparado para manejar los recursos disponibles que derivan de la diversidad cultural de sus estudiantes. Además, un mejor y mayor entendimiento sobre estas diferencias y similitudes culturales permitirá alcanzar los objetivos de inclusión previstos.
- a lo largo de
- equipo
- veces
- tejidos
- a
- se
- Total
- rastrear
- Rastreo
- transferir
- transferido
- tratados
- Tritón
- Twice
- torcedura
- dos
- tipos
- Ultra
- bajo
- único
- Universal
- universidad
- equipo de Manejo Integrado de Plagas de la Universidad de California
- Universidad de Pensilvania
- hasta
- utilizan el
- usado
- usando
- utilizar
- Utilizando
- validado
- Valores
- Variante
- diversos
- versión
- Versus
- vía
- Ver
- virales
- virus
- virus
- vivo
- volúmenes
- fue
- Washington
- Agua
- we
- semana
- Semanas
- peso
- WELL
- Wells
- tuvieron
- que
- todo
- extendido
- dentro de
- mundo
- X
- años
- Rendimiento
- los rendimientos
- Zen
- zephyrnet