Food and Drug Administration (FDA) spelar en avgörande roll för att skydda folkhälsan genom reglering av medicintekniska produkter, för att säkerställa att de är både säkra och effektiva för sin avsedda användning. I samtida tider har ökningen av anslutna enheter fört cybersäkerhet i förgrunden för FDA:s oro; i detta sammanhang är det viktigt att definiera specifika krav för definitionen av en modell för cybersäkerhetshot. I takt med att medicinsk utrustning blir allt mer integrerad med tekniken är behovet av robusta cybersäkerhetsåtgärder för att skydda patientinformation och säkerställa funktionaliteten hos dessa apparater mer kritiskt än någonsin. Efterlevnad av FDA-föreskrifter är inte bara avgörande för tillverkarnas överensstämmelse utan också för säkerheten och förtroendet hos patienter och användare.
Vi har diskuterat digital medicinteknisk utrustning och relaterade krav flera gånger på QualityMedDev-webbplatser, som täcker olika ämnen som t.ex. AI inom medicintekniska produkter, digitala hälsoprodukteroch TGA tillvägagångssätt för reglering av digital medicinteknisk utrustning. Samtidigt har FDA publicerat olika riktlinjer i förhållande till cybersäkerhet, antingen i förhållande till krav före marknaden och eftermarknadskrav.
FDA har fastställt krav på cybersäkerhet som en del av deras regulatoriska tillsyn för att skydda patienter och säkerställa integriteten hos medicinsk utrustning. Dessa standarder gäller för en enhets hela livscykel, från initial design och utveckling till driftsättning och underhåll. Med medicinsk utrustning som blir smartare och mer uppkopplad blir de allt mer sårbara för cyberhot. Integreringen av cybersäkerhetspraxis under utvecklingsfasen av medicinsk utrustning är ett avgörande steg för att mildra riskerna med cyberattacker.
Nyckelkomponenter i FDA:s ramverk för cybersäkerhet
För att säkerställa cybersäkerhetsintegriteten för medicinsk utrustning har FDA beskrivit krav före marknaden som inkluderar behovet för tillverkare att införliva cybersäkerhetshotmodell och säkerhetskontroller från de tidigaste stadierna av enhetens utformning.
Tillverkare av produkter ska upprätta och följa kvalitetssystem för att säkerställa konsekvent överensstämmelse med tillämpliga krav och specifikationer för deras produkter. Kraven för dessa kvalitetssystem finns i Quality System (QS)-förordningen i 21 CFR Part 820 om enheten säljs i USA, eller andra regler för andra länder (t.ex. EU MDR 2017/745 för Europeiska unionen).
Beroende på enhetens karaktär kan QS-kraven vara relevanta under förmarknadsstadiet, eftermarknadsstadiet eller bådadera. I samband med förmarknadsfasen kan att visa rimlig säkerhet om säkerhet och effektivitet för vissa enheter med cybersäkerhetsrisker inbegripa inkludering av dokumentation som är relaterad till QS-förordningen som en del av inlämningen för marknaden. Till exempel kräver ISO 13485:2016 och 21 CFR 820 att tillverkare av alla klasser av enheter som är automatiserade med programvara upprättar procedurer för att kontrollera enhetens design, vilket säkerställer överensstämmelse med specificerade designkrav (kallas "designkontroller"). Inom konstruktionskontroller är tillverkare skyldiga att "etablera och underhålla procedurer för att validera enhetsdesignen", vilket omfattar "programvaruvalidering och riskanalys, där så är lämpligt". Som en del av den obligatoriska programvaruvalideringen och riskanalysen kan tillverkare av mjukvaruenheter behöva införa riskhantering och valideringsprocesser för cybersäkerhet när det är tillämpligt.
Programvaruvalidering och riskhantering utgör avgörande inslag i cybersäkerhetsanalyser, som avgör om en enhet ger rimlig garanti för säkerhet och effektivitet. FDA ger tillverkare mandat att införliva utvecklingsprocesser som beaktar och hanterar programvarurisker under design- och utvecklingsstadierna som en del av designkontrollerna. Dessa processer bör omfatta cybersäkerhetsöverväganden. Detta inkluderar att ta itu med identifieringen av säkerhetsrisker, fastställa designkrav för att kontrollera dessa risker och tillhandahålla bevis för att kontrollerna fungerar som avsett och är effektiva i enhetens avsedda miljö, vilket säkerställer tillräckliga säkerhetsåtgärder.
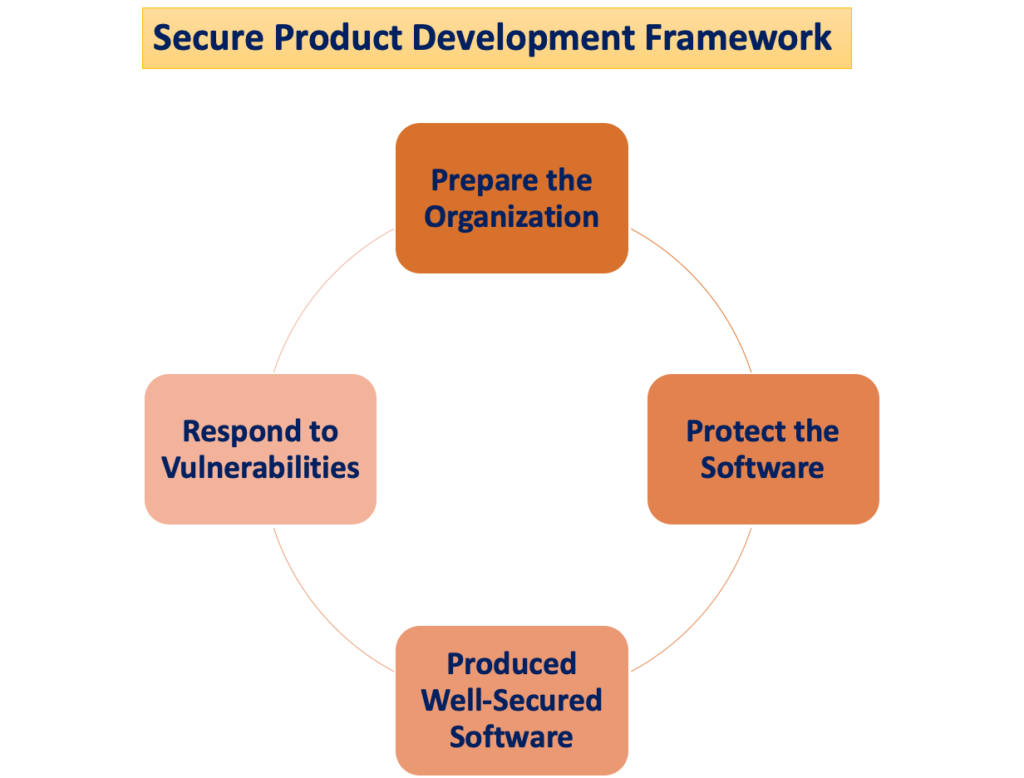
Den "Säkert ramverk för produktutveckling"
Potentialen för patientskador uppstår när cybersäkerhetshot utnyttjar sårbarheter i ett system, och hur lätt dessa hot kan äventyra säkerheten och effektiviteten hos en medicinsk utrustning ökar med antalet identifierade sårbarheter över tiden. A Secure Product Development Framework (SPDF) består av processer som syftar till att identifiera och minska mängden och allvaret av sårbarheter i produkter. Omfattar alla stadier av en produkts livscykel—design, utveckling, release, support och avveckling—SPDF är integrerad.
Under enhetsdesign kan inkorporering av SPDF-processer förhindra behovet av omarbetning när man integrerar anslutningsbaserade funktioner efter marknadsföring eller tar itu med sårbarheter som utgör okontrollerade risker. Möjlig integration med befintliga processer för produkt- och mjukvaruutveckling, riskhantering och det bredare kvalitetssystemet bidrar till SPDF:s mångsidighet.
För att säkerställa överensstämmelse med kvalitetssystemet (QS)-förordningen rekommenderas användning av en SPDF. FDA uppmuntrar tillverkare att anamma SPDF för dess fördelar när det gäller att uppfylla både QS-förordningen och kraven på cybersäkerhet. Det är dock erkänt att alternativa tillvägagångssätt också kan uppfylla QS-förordningen.
Hantering av cybersäkerhetsrisker
Det primära målet med att använda en SPDF (Secure Product Development Framework) är att skapa och underhålla enheter som är både säkra och effektiva. Ur ett säkerhetsperspektiv tjänar dessa enheter också pålitlighet och motståndskraft. Tillverkare och/eller användare (t.ex. patienter, sjukvårdsinrättningar) kan sedan hantera dessa enheter, inklusive installation, konfiguration, uppdateringar och granskning av enhetsloggar, genom enhetsdesignen och tillhörande märkning.
Sjukvårdsinrättningar har möjlighet att hantera dessa enheter inom sina egna ramar för hantering av cybersäkerhetsrisk, såsom det allmänt erkända National Institute of Standards and Technology (NIST) Framework for Improving Critical Infrastructure Cybersecurity, vanligen kallad NIST Cybersecurity Framework eller NIST CSF.
FDA rekommenderar att tillverkare införlivar enhetsdesignprocesser, såsom de som beskrivs i Quality System (QS)-förordningen, för att stärka säker produktutveckling och underhåll. Samtidigt som tillverkarna bibehåller flexibiliteten, kan de också utforska alternativa ramverk som är i linje med QS-förordningen och följer FDA:s rekommendationer för implementering av en SPDF. Exempel inkluderar det medicintekniska specifika ramverket i Medical Device and Health IT Joint Security Plan (JSP) 30 och IEC 81001-5-1. Ramar från andra sektorer, såsom ANSI/ISA 62443-4-1 Säkerhet för industriell automation och kontrollsystem Del 4-1: Produktsäkerhetsutveckling livscykelkrav, kan också uppfylla QS-regler.
I de följande avsnitten av den här artikeln ges rekommendationer för att använda SPDF-processer, som allmänt uppfattas av FDA, som lyfter fram avgörande överväganden för att utveckla enheter som är säkra och effektiva. Dessa processer kompletterar QS-förordningen, och FDA föreslår att tillverkare inkluderar motsvarande dokumentation för granskning i premarket-inlämningar.
Cybersäkerhetshotmodell
Hotmodellering involverar en systematisk process för att identifiera säkerhetsmål, risker och sårbarheter i hela det medicintekniska systemet. Därefter innebär det att man definierar motåtgärder för att förhindra, mildra, övervaka eller reagera på effekterna av hot under hela livscykeln för det medicintekniska systemet. När den tillämpas på lämpligt och heltäckande sätt, fungerar den som grunden för att optimera säkerheten mellan systemkomponenter, produkter, nätverk, applikationer och anslutningar.
När det gäller säkerhetsriskhantering, och för att lokalisera lämpliga säkerhetsrisker och kontroller för det medicinska utrustningssystemet, förespråkar FDA för implementering av hotmodellering för att informera och stödja riskanalysaktiviteter. I samband med riskbedömning föreslår FDA att man integrerar hotmodellering genom hela designprocessen, som omfattar alla delar av det medicintekniska systemet.
Nyckelaspekter av hotmodellen bör omfatta identifiering av risker och begränsningar, informera både före och efter begränsningsrisker som beaktas i cybersäkerhetsriskbedömningen. Dessutom bör modellen artikulera antaganden om det medicintekniska systemet eller användningsmiljön, som att anta att sjukhusnätverk i sig är fientliga. Detta antagande uppmanar tillverkare att överväga scenarier där en motståndare kontrollerar nätverket och kan ändra, släppa och spela upp paket. Dessutom bör hotmodellen fånga cybersäkerhetsrisker som introduceras genom försörjningskedjan, tillverkning, driftsättning, interoperation med andra enheter, underhålls-/uppdateringsaktiviteter och avvecklingsaktiviteter, som kan förbises i en traditionell säkerhetsriskbedömningsprocess.
För inlämningar på marknaden rekommenderar FDA att inkludera hotmodelleringsdokumentation för att visa upp analysen av det medicintekniska systemet, identifiera potentiella säkerhetsrisker som kan påverka säkerhet och effektivitet. Tillverkare har flexibiliteten att välja mellan olika metoder eller kombinationer av metoder för hotmodellering, och motiveringen för deras valda metoder bör bifogas dokumentationen.
Det rekommenderas att utföra hotmodelleringsaktiviteter under designgranskningar, och dokumentationen bör ge tillräcklig information för att FDA ska kunna bedöma och granska säkerhetsfunktionerna som är integrerade i enheten. Denna holistiska utvärdering bör ta hänsyn till både enheten och det bredare system som den fungerar i, och betona enhetens säkerhet och effektivitet.
Huvudelementen i modellerna för cybersäkerhetshot kan sammanfattas enligt följande:
Identifiera potentiella hot
Utvecklingen av en hotmodell fungerar som ett grundläggande steg i cybersäkerhetsprocessen, vilket gör det möjligt för tillverkare att identifiera och förstå potentiella cybersäkerhetshot som är specifika för deras medicinska utrustning. Utvecklingen av en hotmodell fungerar som ett grundläggande steg i cybersäkerhetsprocessen, vilket gör det möjligt för tillverkare att identifiera och förstå potentiella cybersäkerhetshot som är specifika för deras medicinska utrustning. Utvecklingen av en hotmodell fungerar som ett grundläggande steg i cybersäkerhetsprocessen, vilket gör det möjligt för tillverkare att identifiera och förstå potentiella cybersäkerhetshot som är specifika för deras medicinska utrustning.
Riskbedömning och hantering
Riskbedömning och riskhantering innebär att utvärdera de identifierade hoten för att fastställa deras potentiella inverkan och utarbeta lämpliga strategier för att effektivt mildra dessa risker.
Implementering av säkerhetskontroller
Säkerhetskontroller är de skyddsåtgärder eller motåtgärder som implementeras för att skydda den medicinska produktens konfidentialitet, integritet och tillgänglighet från identifierade hot.
Framtida trender i FDA:s riktlinjer för cybersäkerhet
I takt med att hot och teknik utvecklas kommer också FDA:s riktlinjer för cybersäkerhet att utvecklas. Det är viktigt för tillverkarna att förbli informerade och smidiga inför dessa förändringar. Tillverkare bör förutse uppdateringar av regelverk genom att hålla sig à jour med branschtrender och upprätthålla ett proaktivt förhållningssätt till cybersäkerhet.
Förberedelse för oförutsedda utmaningar är nyckeln; Att etablera robusta säkerhetsprotokoll och framåtblickande strategier kommer att positionera tillverkarna för att effektivt reagera på det framtida tillståndet för cybersäkerhetsbestämmelser.
Cybersäkerhet är en aspekt av reglering av medicintekniska produkter som inte kan överskattas – den är lika inneboende för enhetssäkerhet som alla mekaniska eller elektriska egenskaper. FDA har insett detta och genom att implementera ett omfattande ramverk för cybersäkerhet strävar man efter att hålla jämna steg med innovation och samtidigt skydda folkhälsan. Tillverkare, vårdpersonal och patienter måste alla samarbeta för att upprätthålla dessa standarder och säkerställa fortsatt tillförlitlighet och säkerhet för medicinsk utrustning inför cyberhot.
Prenumerera på QualityMedDevs nyhetsbrev
QualityMedDev är en onlineplattform fokuserad på kvalitets- och regleringsämnen för medicintekniska företag; Följ oss på LinkedIn och Twitter för att hålla dig uppdaterad med de viktigaste nyheterna om regleringsområdet.
QualityMedDev är en av de största onlineplattformarna som stödjer verksamhet inom medicintekniska produkter för frågor som rör regelefterlevnad. Vi tillhandahåller regulatoriska konsulttjänster över ett brett spektrum av ämnen, från EU MDR & IVDR till ISO 13485 , inklusive riskhantering, biokompatibilitet, användbarhet och mjukvaruverifiering och validering och i allmänhet stöd vid förberedelse av teknisk dokumentation för MDR.
Vår systerplattform QualityMedDev Academy ger möjligheten att följa onlinekurser och kurser i egen takt fokuserade på ämnen för efterlevnad av regelverk för medicinsk utrustning. Dessa utbildningar, utvecklade i samarbete med högutbildade yrkesverksamma inom medicintekniksektorn, låter dig exponentiellt öka dina kompetenser över ett brett utbud av kvalitets- och regleringsämnen för medicinteknisk verksamhet.

Tveka inte att prenumerera på vårt nyhetsbrev!
- SEO-drivet innehåll och PR-distribution. Bli förstärkt idag.
- PlatoData.Network Vertical Generative Ai. Styrka dig själv. Tillgång här.
- PlatoAiStream. Web3 Intelligence. Kunskap förstärkt. Tillgång här.
- Platoesg. Kol, CleanTech, Energi, Miljö, Sol, Avfallshantering. Tillgång här.
- PlatoHealth. Biotech och kliniska prövningar Intelligence. Tillgång här.
- Källa: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- : har
- :är
- :inte
- :var
- $UPP
- 2016
- 30
- 350
- 820
- 9
- a
- Om oss
- Academy
- medgav
- tvärs
- aktiviteter
- Dessutom
- adress
- adresse
- Lägger
- anslutit sig
- administrering
- rådde
- förespråkar
- smidig
- syftar
- Syftet
- rikta
- Alla
- tillåter
- också
- alternativ
- an
- analyser
- analys
- och
- förutse
- vilken som helst
- tillämplig
- tillämpningar
- tillämpas
- Ansök
- tillvägagångssätt
- tillvägagångssätt
- lämpligt
- lämpligt
- ÄR
- Artikeln
- AS
- aspekt
- aspekter
- bedöma
- bedömning
- associerad
- Antagandet
- antaganden
- försäkran
- At
- Attacker
- Automatiserad
- Automation
- tillgänglighet
- BE
- blir
- passande
- varit
- Fördelarna
- stödja
- båda
- bred
- bredare
- fört
- företag
- affärsverksamhet
- men
- by
- KAN
- kan inte
- kapabel
- fånga
- vissa
- kedja
- utmaningar
- Förändringar
- Välja
- valda
- klasser
- samarbeta
- samverkan
- COM
- kombinationer
- vanligen
- Komplement
- Efterlevnad
- komponenter
- omfattande
- kompromiss
- befruktning
- oro
- konfidentialitet
- konfiguration
- anslutna
- anslutna enheter
- Anslutningar
- Tänk
- överväganden
- anses
- konsekvent
- består
- utgöra
- rådgivning
- samtida
- sammanhang
- fortsatte
- kontroll
- styrning
- kontroller
- Motsvarande
- kunde
- länder
- kurser
- beläggning
- skapa
- kritisk
- Kritisk infrastruktur
- avgörande
- cyber
- Cyberattack
- Cybersäkerhet
- Datum
- definierande
- definition
- demonstrera
- utplacering
- Designa
- designprocessen
- betecknad
- bestämmande
- utvecklade
- utveckla
- Utveckling
- anordning
- enheter
- olika
- digital
- minskande
- diskutera
- dokumentation
- Drop
- drog
- under
- e
- tidigast
- tjänar
- lätta
- Effektiv
- effektivt
- effektivitet
- antingen
- element
- omfamna
- betona
- utnyttjande
- möjliggör
- omfatta
- omfattar
- encompassing
- uppmuntrar
- säkerställa
- säkerställa
- Miljö
- väsentlig
- etablera
- etablerade
- upprättandet
- Eter (ETH)
- EU
- Giltigt körkort
- europeisk union
- utvärdering
- utvärdering
- NÅGONSIN
- bevis
- utvecklas
- exempel
- befintliga
- Exploit
- utforska
- exponentiellt
- Ansikte
- anläggningar
- FDA
- möjlig
- Leverans
- Funktioner
- fält
- Flexibilitet
- fokuserade
- följer
- efter
- följer
- livsmedelsproduktion
- Food and Drug Administration
- Food and Drug Administration (FDA)
- För
- förgrunden
- framåtblickande
- hittade
- fundament
- foundational
- Ramverk
- ramar
- från
- full
- funktionalitet
- Vidare
- framtida
- Allmänt
- allmänhet
- riktlinjer
- skada
- Har
- Hälsa
- hälso-och sjukvård
- Hög
- Markera
- höggradigt
- helhetssyn
- Sjukhuset
- Men
- html
- HTTPS
- Identifiering
- identifierade
- identifiera
- identifiera
- if
- Inverkan
- Konsekvenser
- genomförande
- genomföras
- genomföra
- med Esport
- förbättra
- in
- innefattar
- innefattar
- Inklusive
- införliva
- införlivande
- Öka
- Ökar
- alltmer
- industriell
- industriell automation
- industrin
- underrätta
- informationen
- informeras
- Infrastruktur
- inneboende
- inledande
- Innovation
- Installationen
- exempel
- Institute
- integrerad
- integrerade
- Integrera
- integrering
- integritet
- avsedd
- in
- inneboende
- introducerade
- engagera
- innebär
- ISO
- IT
- DESS
- gemensam
- Ha kvar
- Nyckel
- märkning
- största
- livscykel
- livscykel
- MailChimp
- Huvudsida
- bibehålla
- upprätthålla
- underhåll
- hantera
- ledning
- mandat
- Tillverkare
- Produktion
- max-bredd
- Maj..
- MDR
- åtgärder
- mekanisk
- medicinsk
- medicinsk utrustning
- medicintekniska produkter
- Möt
- möte
- metoder
- metoder
- kanske
- Mildra
- förmildrande
- mildra risker
- modell
- modellering
- modeller
- Övervaka
- mer
- mest
- måste
- nationell
- Natur
- Navigering
- nödvändighet
- Behöver
- nät
- nätverk
- nyheter
- NIST
- antal
- mål
- mål
- of
- on
- ONE
- nätet
- endast
- driva
- fungerar
- Verksamhet
- optimera
- Alternativet
- or
- Övriga
- vår
- skisse
- utgångar
- över
- Tillsyn
- egen
- Fred
- paket
- del
- Patienten
- patienter
- uppfattas
- perspektiv
- fas
- svängbara
- Planen
- plattform
- plato
- Platon Data Intelligence
- PlatonData
- spelar
- plugin
- pose
- placera
- Möjligheten
- inlägg
- potentiell
- praxis
- beredning
- förhindra
- primär
- Proaktiv
- förfaranden
- process
- processer
- Produkt
- produktutveckling
- Produkter
- yrkesmän/kvinnor
- prompter
- skydda
- skydda
- protokoll
- ge
- förutsatt
- ger
- tillhandahålla
- allmän
- folkhälsan
- publicerade
- kvalitet
- mängd
- område
- logiska grunden
- rimlig
- erkänt
- rekommendationer
- rekommenderas
- rekommenderar
- avses
- reglering
- föreskrifter
- regulatorer
- Regelefterlevnad
- relaterad
- förhållande
- frigöra
- tillförlitlighet
- förblir
- Obligatorisk
- Krav
- motståndskraft
- Svara
- översyn
- Omdömen
- Rise
- Risk
- riskbedömning
- riskhanterings
- risker
- robusta
- Roll
- säker
- skydd
- skyddsåtgärder
- Säkerhet
- Samma
- scenarier
- sektioner
- sektor
- Sektorer
- säkra
- säkerhet
- Säkerhetsåtgärder
- hantering av säkerhetsrisker
- säkerhetsrisker
- serverar
- flera
- stränghet
- skall
- visa
- syster
- skicklig
- smartare
- So
- Mjukvara
- mjukvaruutveckling
- säljs
- specifik
- specifikationer
- specificerade
- Etapp
- stadier
- standarder
- Ange
- Stater
- bo
- vistas
- Steg
- strategier
- underkastelse
- Inlagor
- prenumerera
- Senare
- sådana
- tillräcklig
- Föreslår
- lämplig
- leverera
- leveranskedjan
- stödja
- Stödjande
- system
- System
- Teknisk
- Teknologi
- än
- den där
- Smakämnen
- Framtiden
- deras
- sedan
- Dessa
- de
- detta
- de
- hot
- hot
- Genom
- hela
- tid
- gånger
- till
- alltför
- ämnen
- traditionell
- Utbildning
- Trender
- Litar
- trovärdighet
- förstå
- oförutsedd
- fackliga
- United
- USA
- Uppdateringar
- upprätthålla
- URL
- us
- användbarhet
- Användning
- användning
- användare
- Använda
- validera
- godkännande
- olika
- Verifiering
- mångsidighet
- sårbarheter
- Sårbara
- we
- webbsidor
- när
- om
- som
- medan
- brett
- kommer
- med
- inom
- Wordpress
- WordPress plugin
- dig
- Din
- zephyrnet





