Todos os experimentos e procedimentos foram aprovados pelos conselhos e comitês reguladores locais e foram obrigados a cumprir os protocolos do estudo. Todos os procedimentos com ratos foram realizados no Caltech, aprovados pelo Comitê Institucional de Cuidados e Uso de Animais da Caltech (IACUC; protocolo 1738). Os procedimentos com saguis (protocolo TGC-03) e macacos adultos (protocolo LN-14) ocorreram no NIH e foram aprovados pelo NIH IACUC. Os procedimentos do sagui também foram concluídos na Universidade da Califórnia em San Diego (UCSD) (protocolo S09147) e estavam em conformidade e aprovados pela UCSD IACUC. Os procedimentos com macacos infantis foram realizados no Centro Nacional de Pesquisa de Primatas da Califórnia, na Universidade da Califórnia Davis, e foram aprovados pelo IACUC local (protocolo 22525). Os procedimentos do macaco verde ocorreram em Virscio e foram aprovados pelo IACUC local.
Geração de biblioteca de DNA AAV
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jy89g2w/v1). Inicialmente geramos diversidade no nível do DNA, que então usamos para produzir material de transfecção para produzir a biblioteca do capsídeo AAV, conforme descrito anteriormente em detalhes16. Para a biblioteca de primeira rodada, introduzimos esta diversidade genética usando primers contendo nucleotídeos degenerados inseridos entre os aminoácidos 588 e 589 (refs. 12,13,16) (numeração VP1; Figura Complementar. 1a). Usamos um primer reverso contendo 7 nucleotídeos degenerados ([NNK] × 7) para gerar aleatoriamente fragmentos de reação em cadeia da polimerase (PCR) contendo sequências 7mer únicas inseridas no boné gene. Para a biblioteca de DNA de segunda rodada, usamos um pool de oligos sintéticos (Twist Bioscience) como primer reverso, codificando apenas variantes selecionadas para triagem adicional (total, 66,628 oligos de DNA; 33,314 variantes recuperadas após seleções de primeira rodada mais um códon modificado réplica de cada um). Todos os primers reversos continham uma saliência 20' de 5 pb complementar ao boné sequência próxima à sequência da enzima de restrição AgeI e foram emparelhados com um primer forward contendo uma saliência 20 'de 5 pb perto da sequência da enzima de restrição XbaI. Em seguida, inserimos os fragmentos de PCR contendo a região diversificada no plasmídeo rAAV-ΔCAP-in-cis-Lox via montagem Gibson para gerar a biblioteca de DNA de AAV resultante, ou seja, rAAV-CAP-in-cis-Lox, usando NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, E2621).
Produção de biblioteca de capsídeo AAV
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5jyl8jyz9g2w/v1). Geramos bibliotecas de capsídeos AAV de acordo com protocolos publicados anteriormente16,70. Resumidamente, transfectamos células HEK293T (ATCC, cat # CRL-3216; RRID: CVCL_0063) em placas de cultura de tecidos de 150 mm usando polietilenimina linear de grau de transfecção (PEI; Polysciences). Em cada placa, transfectamos quatro plasmídeos: (1) a biblioteca de DNA de AAV rAAV-Cap-in-cis-Lox montada, que é flanqueada por repetições terminais invertidas necessárias para a encapsidação de AAV; (2) AAV2/9 REP-AAP-ΔCAP, que codifica as proteínas suplementares REP e AAP necessárias para a produção de AAV com o terminal C do boné gene excisado para evitar a recombinação com a biblioteca de DNA de AAV e subsequente produção de AAV competente para replicação; (3) pHelper, que codifica as proteínas adenovirais necessárias para a produção de AAV; e (4) pUC18 (Addgene ID: 50004; RRID: Addgene_50004), que não contém vetor de expressão de mamífero, mas é usado como DNA de enchimento para atingir a proporção apropriada de nitrogênio para fosfato para transfecção ideal de PEI. Durante a preparação da mistura PEI-DNA, adicionamos 10 ng de nossa biblioteca de DNA AAV (rAAV-Cap-in-cis-Lox) para cada placa de 150 mm e combinamos AAV2/9 REP-AAP-ΔCAP, pUC18 e pHelper em um Proporção 1:1:2 (40 µg de DNA total por placa de 150 mm). Às 60 horas pós-transfecção, purificamos a biblioteca do capsídeo de AAV tanto do sedimento celular quanto do meio usando precipitação com polietilenoglicol e ultracentrifugação com gradiente de iodixanol. Usando PCR quantitativo, determinamos então o título das bibliotecas do capsídeo de AAV amplificando genomas virais resistentes a DNaseI em relação a um padrão de genoma linearizado de acordo com protocolos estabelecidos70.
Experimentos com saguis
Seleções da biblioteca Capsid
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Todo o sagui (C.jacchus) os procedimentos foram realizados no Instituto Nacional de Saúde Mental (NIMH) e aprovados pelo IACUC local. Os saguis nasceram e foram criados em colônias do NIMH e alojados em grupos familiares sob condições padrão de 27 °C e 50% de umidade. Eles foram alimentados ad libitum e receberam enriquecimento como parte do programa de enriquecimento de primatas para NHPs no NIH. Para todos os saguis utilizados neste estudo, não houve anticorpos neutralizantes detectáveis numa diluição de soro de 1:5 antes das infusões intravenosas (ensaiadas pelo Penn Vector Core, Universidade da Pensilvânia). Eles foram então alojados individualmente por vários dias e aclimatados em uma nova sala antes das injeções. Quatro homens adultos foram utilizados para a triagem da biblioteca, dois de cada para as bibliotecas de primeira e segunda rodada. No dia anterior à infusão, a alimentação dos animais foi retirada. Os animais foram anestesiados com isoflurano em oxigênio, a pele sobre a veia femoral foi raspada e higienizada com esfoliante de isopropanol e 2 × 1012 vg da biblioteca de capsídeo de AAV foi infundida durante vários minutos. A anestesia foi suspensa e os animais foram monitorados até se tornarem ativos, quando foram devolvidos às suas gaiolas. A atividade e o comportamento foram monitorados de perto durante os 3 dias seguintes, com observações diárias a partir de então.
Às 4 semanas após a injeção, os saguis foram eutanasiados (Euthanasia, VetOne) e perfundidos com 1x solução salina tamponada com fosfato (PBS). Após a primeira rodada da biblioteca, o cérebro foi cortado em quatro blocos coronais, congelado rapidamente em 2-metilbutano (Sigma-Aldrich, M32631), resfriado com gelo seco e armazenado a -80°C para armazenamento a longo prazo. Após a segunda rodada da biblioteca, o cérebro foi cortado em seis blocos coronais e, junto com seções da medula espinhal e do fígado, foi congelado rapidamente e armazenado a -80 °C para armazenamento a longo prazo.
Caracterização individual de AAVs em saguis
Os detalhes dos procedimentos nesta seção podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 e https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Dois saguis comuns adultos (C.jacchus) foram utilizados para este experimento: Conan (homem, 2.8 anos, 0.386 kg) e Sandy (mulher, 5.8 anos, 0.468 kg) (Tabela Suplementar 3 fornece mais detalhes). Eles foram alojados em condições padrão de 27°C e 50% de umidade, com acesso ad libitum a comida e água. Todos os animais foram alojados em grupo e os experimentos foram realizados no Laboratório de Sistemas Corticais e Comportamento da UCSD. Todos os experimentos foram aprovados pela UCSD IACUC. No dia anterior à infusão, a alimentação dos animais foi retirada.
Os animais foram anestesiados com cetamina (Ketaset, Zoetis 043-304, 20 mg kg-1), a pele sobre a veia safena foi raspada e higienizada com esfoliante de isopropanol e 2 × 1013 peso corporal-1 de AAV foi infundido durante 5 minutos. Os animais foram monitorados até se tornarem ativos, quando foram devolvidos às suas gaiolas. A atividade e o comportamento foram monitorados de perto durante os 3 dias seguintes, com observações diárias a partir de então. Amostras de sangue foram coletadas nos dias 1, 7, 14, 21 e 31 para medir a concentração viral no plasma.
Aos 31 dias após a injeção, os saguis foram anestesiados com cetamina conforme descrito anteriormente e depois sacrificados (Euthasol, Virbac 200-071, 1 ml kg-1) e perfundido com 1× PBS. Cérebros e órgãos foram cortados ao meio e metade foi congelada rapidamente em 2-metilbutano (Sigma-Aldrich, M32631), resfriada com gelo seco e armazenada a -80 °C. A outra metade foi fixada em paraformaldeído a 4% (PFA) (Thermo Scientific, J19943-K2) durante a noite e depois armazenada a 4 °C em azida PBS (Sigma-Aldrich, S2002-100G, 0.025%). As amostras foram então enviadas ao Instituto de Tecnologia da Califórnia (Caltech) para análise. Para coloração de GLUT1, incubamos fatias com anti-GLUT1 de coelho (1:200; Millipore-Sigma, cat # 07-1401; RRID: AB_1587074), realizamos três a cinco lavagens com PBS, incubamos com IgG anti-coelho de burro (1: 200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) e lavado três a cinco vezes antes da montagem. Diluímos todos os anticorpos e realizamos todas as incubações usando PBS suplementado com 0.1% de Triton X-100 (Sigma-Aldrich, T8787) e 10% de soro de burro normal (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) durante a noite em temperatura ambiente com agitação.
Extração de DNA de biblioteca viral e preparação de amostras NGS
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.bp2l695zklqe/v2). Relatamos anteriormente que o DNA da biblioteca viral e o RNA do hospedeiro endógeno podem ser isolados usando TRIzol precipitando o ácido nucleico da fase aquosa12,16. Portanto, para extrair o DNA da biblioteca viral do tecido de sagui, homogeneizamos 100 mg de medula espinhal, fígado e cada bloco coronal do cérebro em TRIzol (Life Technologies, 15596) usando um BeadBug (Benchmark Scientific, D1036) e isolamos ácidos nucléicos do fase aquosa de acordo com o protocolo recomendado pelo fabricante. Tratamos o precipitado reconstituído com RNase (Invitrogen, AM2288) e digerimos com SmaI para melhorar a recuperação do DNA viral a jusante via PCR. Após a digestão, purificamos com um kit Zymo DNA Clean and Concentrator (D4033) de acordo com o protocolo recomendado pelo fabricante e armazenamos o DNA viral purificado a -20 °C.
Para anexar adaptadores Illumina flanqueando a região diversificada, primeiro amplificamos por PCR a região contendo nossa inserção 7mer usando 50% do DNA viral total extraído como modelo (25 ciclos). Após a purificação do DNA do Zymo, diluímos as amostras a 1:100 e amplificamos ainda mais em torno da região variável com dez ciclos de PCR, anexando regiões de ligação para a próxima reação de PCR. Finalmente, anexamos adaptadores de células de fluxo Illumina e índices exclusivos usando NEBNext Dual Index Primers (New England Biolabs, E7600) por meio de mais dez ciclos de PCR. Em seguida, purificamos em gel os produtos finais de PCR usando um gel de agarose de baixo ponto de fusão a 2% (Thermo Fisher Scientific, 16520050) e recuperamos a banda de 210 pb.
Apenas para a biblioteca de segunda rodada, também isolamos o ssDNA da biblioteca AAV encapsidada para NGS para calcular os escores de enriquecimento da biblioteca, uma métrica quantitativa que usamos para normalizar as diferenças no título das várias variantes em nossa biblioteca (ver ref. 16 e a seção ‘Alinhamento de leitura NGS, análise e geração de gráficos de rede’). Para isolar os genomas virais encapsidados, tratamos a biblioteca do capsídeo AAV com DNaseI e digerimos os capsídeos usando proteinase K. Em seguida, purificamos o ssDNA usando fenol-clorofórmio, amplificamos os transgenes virais por duas etapas de amplificação por PCR para adicionar adaptadores e índices para Illumina NGS e purificamos usando eletroforese em gel. Este DNA da biblioteca viral, juntamente com o DNA viral extraído do tecido, foi enviado para sequenciamento profundo usando um sistema Illumina HiSeq 2500 (Laboratório de Genética e Genômica Millard e Muriel Jacobs, Caltech).
Alinhamento de leitura NGS, análise e geração de gráficos de rede
Arquivos FASTQ brutos de execuções NGS foram processados com scripts personalizados (https://github.com/GradinaruLab/protfarm e https://github.com/GradinaruLab/mCREATE)16. Para a biblioteca de primeira rodada, o pipeline para processar esses conjuntos de dados envolveu filtragem para remover leituras de baixa qualidade, utilizando um índice de qualidade para cada sequência e eliminando preconceitos de mutações induzidas por PCR ou alto conteúdo de GC. O conjunto de dados filtrado foi então alinhado por um algoritmo de correspondência de string perfeita e cortado para melhorar a qualidade do alinhamento. Em seguida, exibimos as contagens absolutas de leitura para cada variante durante a execução de sequenciamento dentro de cada tecido, e todas as 33,314 variantes encontradas no cérebro foram escolhidas para as seleções da segunda rodada.
Após as seleções da segunda rodada, realizamos a mesma análise para exibir a contagem absoluta de leituras variantes da biblioteca de vírus injetados e de cada variante dentro de cada tecido. Além disso, calculamos o enriquecimento da biblioteca16 para cada variante dentro de cada tecido:
$${overline{{rm{RC}}}}_{x,{rm{injetado}},{rm{biblioteca}}}=,frac{{{rm{RC}}}_{x,{rm{ injetado}},{rm{biblioteca}}}}{mathop{sum }nolimits_{i=1}^{{N}_{{rm{injetado}},{rm{biblioteca}}}}{{rm{RC }}}_{i,{rm{injetado},{biblioteca}}}},$$
(1)
$${overline{{rm{RC}}}}_{x,{rm{tecido}}}=,frac{{{rm{RC}}}_{x,{rm{vírus}}}}{mathop {sum }nolimits_{i=1}^{{N}_{{rm{tecido}}}}{{rm{RC}}}_{i,{rm{tecido}}}},$$
(2)
$${rm{Biblioteca},{enriquecimento}}=,{log }_{10}left(frac{{overline{{rm{RC}}}}_{x,rm{{injetado},{biblioteca}} }}{{overline{{rm{RC}}}}_{x,{rm{tissue}}}}direita),$$
(3)
tal que para uma determinada amostra y (por exemplo, a biblioteca de vírus injetada ou uma amostra de tecido), RCx,y é a contagem absoluta de leituras da variante x, Ny é o número total de variantes recuperadas e ({overline{{rm{RC}}}}_{x,{y}}) é a contagem de leitura normalizada.
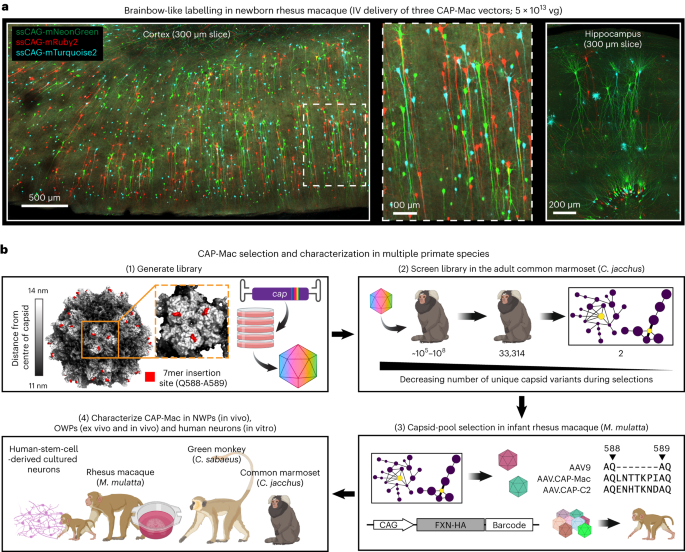
Para construir o gráfico de agrupamento de sequências CAP-Mac, filtramos os dados NGS de segunda rodada com base nos seguintes critérios: (1) ≥100 contagem de leituras na amostra da biblioteca injetada (24,186/33,314 variantes), (2) ≥0.7 enriquecimento da biblioteca pontuação em mais de duas amostras de cérebro (415 variantes) e (3) pelo menos mais duas amostras de cérebro com enriquecimento de biblioteca ≥0.7 do que amostras de cérebro com enriquecimento de biblioteca inferior a -0.7 (323 variantes). Para construir o gráfico de sequência CAP-C2, filtramos os dados NGS da segunda rodada com base nos seguintes critérios: (1) ≥100 contagem de leituras na amostra da biblioteca injetada e (2) ambas as réplicas de códons presentes em pelo menos duas amostras cerebrais com ≥0.7 enriquecimento de biblioteca (95 variantes). Essas variantes foram então processadas independentemente para determinar distâncias de Hamming reversas em pares (https://github.com/GradinaruLab/mCREATE) e agrupado usando Cytoscape (v. 3.9.0; RRID: SCR_003032) conforme descrito anteriormente em detalhes16. As redes apresentadas mostram variantes de capsídeo (nós) conectadas por arestas se a distância de Hamming reversa entre pares for ≥3.
Clonagem de variantes individuais do capsídeo AAV
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.n2bvj87ebgk5/v1). Para caracterização de variante única, clonamos novos plasmídeos variantes digerindo uma versão modificada do backbone pUCmini-iCAP-PHP.eB (Addgene ID: 103005; RRID: Addgene_103005) usando MscI e AgeI. Projetamos um primer de 100 pb que continha a inserção desejada de 21 pb para cada variante do capsídeo e as regiões complementares ao modelo AAV9 com ~ 20 pb sobrepondo a estrutura digerida. Em seguida, montamos o plasmídeo variante usando NEBuilder HiFi DNA Assembly Master Mix, combinando 5 μl de primer 200 nM com 30 ng de estrutura digerida na mistura de reação. O plasmídeo do capsídeo usado para produzir AAV.CAP-Mac está disponível em Addgene (Addgene ID: 200658; RRID: Addgene_200658).
Produção e purificação individual de AAV
Os detalhes deste procedimento podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.14egn2dqzg5d/v1). Para produzir variantes para testes de pool, seguimos nosso protocolo publicado anteriormente70 usando placas de cultura de tecidos de 150 mm. Para caracterização individual de AAV.CAP-Mac e AAV9 in vivo e in vitro, adotamos nosso protocolo publicado para utilizar CellSTACKs de dez camadas (Corning, 3320) para produzir eficientemente vírus em alto título para dosar macacos rhesus e macacos verdes. Especificamente, passamos vinte placas de 150 mm com aproximadamente 70% de confluência em um CellSTACK de dez camadas 24 h antes da transfecção. No dia da transfecção, preparamos a mistura de transfecção PEI-DNA para quarenta placas de 150 mm e combinamos a mistura de transfecção com meio e realizamos uma troca completa de meio para o CellSTACK. Coletamos e trocamos a mídia 72 horas após a transfecção, semelhante à produção em placas de 150 mm. Às 120 horas pós-transfecção, adicionamos ácido etilenodiaminotetracético (Invitrogen, 15575020) a uma concentração final de 10 mM e incubamos a 37 ° C por 20 minutos, ocasionalmente girando e batendo nas laterais do CellSTACK para separar as células. Em seguida, removemos a mistura de mídia e células e prosseguimos com o protocolo de purificação de AAV70. É digno de nota que, durante a etapa de troca de tampão após a ultracentifugação, utilizamos concentradores centrífugos de proteínas com membranas de polietersulfona (Thermo Scientific, 88533) em vez de dispositivos de filtração Amicon e usamos PBS de Dulbecco suplementado com 0.001% de Pluronic F-68 (Gibco, 24040032).
Experimentos com roedores
Todos os procedimentos com roedores foram realizados no Caltech e aprovados pelo IACUC local. Compramos camundongos C57BL/6J (cepa #: 000664; RRID: IMSR_JAX:000664), BALB/cJ (cepa #: 000651; RRID: IMSR_JAX:000651) e DBA/2J (cepa #: 000671; RRID: IMSR_JAX:000671). (todos homens, 6–8 semanas de idade) do Laboratório Jackson. Para administração IV em camundongos, administramos 5 × 1011 vg de vírus através do seio retro-orbital70,71 usando uma seringa de insulina de calibre 31 (BD, 328438). Consulte protocols.io para obter mais detalhes sobre injeções retro-orbitais de AAV em camundongos (https://doi.org/10.17504/protocols.io.3byl4joy8lo5/v1). Para administração intracerebroventricular (ICV) em camundongos, injetamos 5.0 × 1010 ou 1.5 × 1011 vg para o ventrículo lateral. Resumidamente, anestesiamos camundongos usando isoflurano (5% para indução, 1–3% para manutenção) com 95% de O2/ 5% CO2 (1 l min-1) e os camundongos foram fixados com a cabeça em uma estrutura estereotáxica. Após raspar a cabeça e esterilizar a área com clorohexidina, administramos por via subcutânea 0.05 ml de 2.5 mg ml-1 bupivacaína, e uma incisão na linha média foi feita e o crânio foi limpo de sangue e tecido conjuntivo. Após o nivelamento da cabeça, foram perfurados orifícios de trepanação bilateralmente acima dos ventrículos laterais (0.60 mm posterior ao bregma e 1.15 mm da linha média). Os vetores virais foram aspirados em seringas NanoFil de 10 µl (World Precision Instruments) usando uma agulha de microinjeção de calibre 33, e a agulha foi lentamente abaixada no ventrículo lateral (1.6 mm da superfície pial). A agulha foi deixada no lugar por aproximadamente 5 min e 3-5 μl de vetor viral foram injetados usando uma bomba de microseringa (World Precision Instruments, UMP3) e um controlador de bomba (World Precision Instruments, Mircro3) a uma taxa de 300 nl min.-1. Todos os ratos receberam intraoperatoriamente 1 mg kg-1 de buprenorfina SR e 5 mg kg-1 de cetoprofeno por via subcutânea e 30 mg kg-1 de ibuprofeno e 60 mg kg-1 de trimetoprima/sulfametoxazol por 5 dias após a cirurgia. Consulte protocols.io para obter mais detalhes sobre injeções ICV de AAV em camundongos (https://doi.org/10.17504/protocols.io.5qpvorm4dv4o/v1). Após 3 semanas de expressão, todos os ratos foram perfundidos com PBS e fixados em PFA a 4%. Todos os órgãos foram extraídos, incubados em PFA a 4.00% durante a noite, transferidos para PBS suplementado com azida de sódio a 0.01% e armazenados a 4 °C para armazenamento a longo prazo. Cortamos o cérebro em seções de 100 μm com um vibratome (Leica Biosystems, VT1200S), montado em Prolong Diamond Antifade (Invitrogen, P36970) e fotografado usando um microscópio confocal (Zeiss, LSM 880) usando ZEN (Black edition). Consulte protocols.io para obter mais detalhes sobre o manuseio de tecidos (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 e https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1).
Experimentos com macacos Rhesus
Os detalhes dos procedimentos nesta seção podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1). Macacos neonatos (0.45–1.40 kg) foram desmamados ao nascer. No primeiro mês, os macacos foram infundidos com vetores AAV por via intravenosa ou intratecal. Todos os macacos adultos (8–17 anos; 4.65–11 kg) incluídos neste estudo receberam infusão de AAV apenas por administração intravenosa. Para as injeções intravenosas, os animais foram anestesiados com cetamina (0.10 ml) e a pele sobre a veia safena foi raspada e higienizada. AAV (entre 2 × 1013 e 1 × 1014 peso corporal-1) foi infundido lentamente na veia safena durante ~1 min em <0.75 ml de PBS. Para injeções de ICM, os animais receberam um sedativo por via intramuscular e a área da pele no pescoço foi raspada e preparada assepticamente. Uma agulha foi avançada na cisterna magna para remover uma pequena quantidade de LCR proporcional à quantidade de líquido injetado. Em seguida, uma seringa estéril contendo o preparado estéril da AAV (1.5 × 1012 ou 2.5 × 1013 peso corporal-1) proporcional à quantidade de líquido coletado foi fixado assepticamente e injetado lentamente. Todos os animais foram monitorados durante a recuperação da sedação ao longo do dia e depois diariamente para quaisquer resultados adversos. Todos os macacos foram alojados individualmente à vista e ao som de membros da mesma espécie. O tecido foi coletado 4–11 semanas após a injeção. Os animais foram profundamente anestesiados e receberam pentobarbital sódico de acordo com as diretrizes para a eutanásia humana de animais do Centro Nacional de Pesquisa de Primatas da Califórnia. Todo o material injetado em macacos rhesus estava livre de endotoxinas (<0.1 EU ml-1), e a pureza da proteína foi confirmada por eletroforese em gel de dodecil sulfato de sódio-poliacrilamida. Tabelas Suplementares 4 e 5 listar a via de administração, variantes de AAV, dose viral, carga genética e duração da expressão para cada experimento.
Testes de pool em macacos rhesus
Os detalhes dos procedimentos nesta seção podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1, https://doi.org/10.17504/protocols.io.3byl4jo68lo5/v1 e https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Experimentos em pool de macacos neonatos (RM-001 a RM-004) foram realizados no CNPRC da UC Davis e aprovados pelo IACUC local. Experimentos em piscinas de macacos adultos (RMN-001 e RMN-002) foram realizados no NIMH e aprovados pelo IACUC local. Os macacos foram perfundidos com PBS isento de RNase gelado. No momento da perfusão, um hemisfério do cérebro foi congelado rapidamente e o outro hemisfério foi seccionado em blocos coronais de 4 mm e pós-fixado em PFA a 4% por 48 horas e transferido para Caltech para processamento adicional. Para coloração de HA, incubamos fatias com anti-HA de coelho (1:200; Cell Signaling Technology, cat # 3724; RRID: AB_1549585), realizamos três a cinco lavagens com PBS, incubamos com IgG anti-coelho de burro (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) e lavado três a cinco vezes antes da montagem. Diluímos todos os anticorpos e realizamos todas as incubações usando PBS suplementado com 0.1% de Triton X-100 (Sigma-Aldrich, T8787) e 10% de soro de burro normal (Jackson ImmunoResearch Labs, cat # 017-000-121; RRID: AB_2337258) durante a noite à temperatura ambiente com agitação.
Para isolar o DNA viral e o RNA total, fatias de 100 mg do cérebro e do fígado foram homogeneizadas em TRIzol (Life Technologies, 15596) usando um BeadBug (Benchmark Scientific, D1036) e o DNA e RNA total foram recuperados de acordo com o protocolo recomendado pelo fabricante . O DNA recuperado foi tratado com RNase, digerido por restrição com SmaI e purificado com um kit Zymo DNA Clean and Concentrator (D4033). O RNA recuperado foi tratado com DNase e o cDNA foi gerado a partir do mRNA usando SuperScript III (Thermo Fisher Scientific, 18080093) e primers oligo (dT) de acordo com o protocolo recomendado pelo fabricante. Usamos PCR para amplificar a região do código de barras usando 50 ng de DNA viral ou cDNA como modelo. Após a purificação do DNA do Zymo, diluímos as amostras a 1:100 e amplificamos ainda mais a região do código de barras usando primers para anexar adaptadores para Illumina NGS. Após a limpeza, estes produtos foram ainda amplificados utilizando NEBNext Dual Index Primers para sequenciação Illumina (New England Biolabs, E7600) durante dez ciclos. Em seguida, purificamos em gel os produtos finais de PCR usando um gel de agarose de baixo ponto de fusão a 2% (Thermo Fisher Scientific, 16520050). O enriquecimento do teste de pool foi calculado de forma idêntica ao enriquecimento da biblioteca, mas está representado na Fig. 2b, c em uma escala linear.
Caracterização individual de CAP-Mac em macacos rhesus
Os detalhes dos procedimentos nesta seção podem ser encontrados em protocols.io (https://doi.org/10.17504/protocols.io.5qpvormddv4o/v1 e https://doi.org/10.17504/protocols.io.j8nlkwxxwl5r/v1). Macacos neonatos foram perfundidos com PBS e 4% de PFA. O cérebro foi seccionado em blocos coronais de 4 mm e todo o tecido foi pós-fixado em PFA a 4% por 3 dias antes do armazenamento em PBS. O único macaco adulto usado para caracterização individual in vivo (RM-020; 17 anos, 11 kg) foi perfundido com PBS livre de RNase, e um meio hemisfério foi congelado rapidamente e o outro seccionado em blocos coronais de 4 mm e pós-fixado em 4% PFA. Todo o tecido foi transferido para Caltech para processamento posterior. Cérebros e fígados foram seccionados em fatias de 100 μm usando um vibratome. Além disso, seções do cérebro e da medula espinhal foram incubadas em sacarose a 30% durante a noite e embebidas em O.C.T. composto (Scigen, 4586) e seccionado em fatias de 50 μm usando um criostato (Leica Biosystems, CM1950). Todas as fatias foram montadas usando Prolong Diamond Antifade e fotografadas usando um microscópio confocal. Para coloração GFP da medula espinhal e fatias cerebrais do macaco administrado por via intratecal, incubamos fatias com anti-GFP de frango (1:500; Aves Labs, cat # GFP-1020; RRID: AB_10000240), realizamos três a cinco lavagens com PBS , incubado com IgY anti-frango de burro (1:200; Jackson ImmunoResearch Lab, cat # 703-605-155; RRID: AB_2340379) e lavado três a cinco vezes antes da montagem. Diluímos todos os anticorpos e realizamos todas as incubações utilizando PBS suplementado com 0.1% de Triton X-100 (Sigma-Aldrich, T8787) e 10% de soro de burro normal (Jackson ImmunoResearch, 017-000-121) durante a noite à temperatura ambiente com agitação.
Para reconstrução morfológica, seccionamos cérebros em seções de 300 μm e os incubamos em uma solução correspondente ao índice de refração72 por 72 horas antes da montagem em uma lâmina imersa na solução de correspondência de índice de refração. Fizemos imagens usando um microscópio confocal e objetiva × 25 (LD LCI Plan-Apochromat × 25 / 0.8 Imm Corr DIC) usando 100% de glicerol como fluido de imersão. Nós capturamos azulejos Z pilhas (1,024 × 1,024 para cada quadro usando as configurações de captura sugeridas) em torno das células de interesse e campos de visão apropriados cortados para rastreamento. O rastreamento foi feito em Imaris (Oxford Instruments; RRID: SCR_007370) utilizando métodos semiautomáticos e automatizados.
Para quantificação de neurônios (NeuN) e astrócitos (S100β), as fatias foram coradas usando anticorpo anti-NeuN (EPR12763) (1:200; Abcam, cat # ab177487; RRID: AB_2532109) ou anticorpo anti-S100β (1:200; Abcam , cat # ab52642; RRID: AB_882426) durante a noite em PBS suplementado com 0.1% de Triton X-100 e 10% de soro de burro normal. As fatias foram lavadas três a cinco vezes com PBS e incubadas durante a noite em anticorpo IgG anti-coelho conjugado com Alexa Fluor 647 (1:200; Jackson ImmunoResearch Labs, cat # 711-605-152; RRID: AB_2492288) em PBS + 0.1%. Triton X-100 + 10% de soro de burro normal. Após três a cinco lavagens e montagem com Prolong Diamond Antifade, obtivemos Z pilhas usando um microscópio confocal e uma objetiva ×25. Segmentamos células positivas para NeuN e XFP usando scripts personalizados em Python (RRID: SCR_008394) e Cellpose (https://www.cellpose.org/; RRID: SCR_021716)73.
Imagem ex vivo de dois fótons
Fatias cerebrais de tamanhos adequados para imagens foram preparadas com uma espessura de 400 µm a partir de fatias maiores usando um vibratome e armazenadas em líquido cefalorraquidiano artificial borbulhado com gás carbógeno antes da imagem de dois fótons, conforme descrito anteriormente em protocolos publicados74,75. Para testar as respostas do GCaMP8s, a estimulação elétrica (4–5 V, 80 Hz, 0.3 s de duração) com o número indicado de pulsos foi aplicada usando um eletrodo monopolar extracelular colocado a 100–200 µm de distância do neurônio fotografado. A taxa de quadros da imagem foi de 30 Hz. Traços de regiões segmentadas de interesse foram plotados como ΔF/F0 = (F(t) - F0)/F0, Onde F0 é definido como a média de todos os valores de fluorescência antes da estimulação elétrica. O tempo de subida foi definido como o tempo necessário para a fase ascendente do sinal atingir de 10% do pico a 90% do pico. A constante de tempo de decaimento foi obtida ajustando as somas das exponenciais à fase de decaimento do sinal. A relação sinal-ruído foi obtida dividindo a amplitude do pico do sinal pelo desvio padrão do traço de fluorescência antes da estimulação elétrica.
Caracterização em fatia de macaco rhesus adulto
Um macaco rhesus adulto (14 anos e 1 mês de idade; 10.83 kg) do Centro Nacional de Pesquisa de Primatas de Washington foi planejado para eutanásia de rotina, e o cérebro foi coletado como parte do Programa de Distribuição de Tecidos da instalação. Um bloco do giro temporal superior foi seccionado em fatias de 300 μm e as fatias foram recuperadas74 e cultivado em uma interface de membrana ar-líquido, como descrito anteriormente76. Aproximadamente 30 min após o plaqueamento das fatias, administramos 1–2 μl de AAV (5 × 1013 vgml-1 de embalagem AAV9 ou AAV.CAP-Mac ssCAG-FXN-HA ou ssCAG-eGFP). Os experimentos foram realizados em triplicatas biológicas para cada condição e o meio de cultura foi atualizado a cada 48 horas até a coleta do tecido, 8 dias após a transdução. No dia da coleta de tecido, as fatias foram fotografadas para confirmar a transdução, as fatias foram cortadas ao meio e cada meia fatia foi congelada rapidamente em banho de gelo seco-etanol. As amostras foram armazenadas a -20 °C até processamento adicional.
Cada meia fatia foi processada (uma para recuperação de DNA e uma para recuperação de RNA). O DNA foi isolado usando o kit Qiagen DNeasy Blood and Tissue (Qiagen, catálogo # 69504) e o RNA foi recuperado usando TRIzol (Thermo Fisher Scientific, catálogo # 15596026) e o kit PureLink RNA Mini (Thermo Fisher Scientific, catálogo # 12183018A). O DNA foi removido da amostra de RNA modificando a primeira lavagem do kit PureLink RNA Mini da seguinte forma: lavar com 350 µl de tampão de lavagem 1, depois adicionar 80 µl de DNaseI livre de RNase em tampão RDD (catálogo Qiagen # 79254) e incubar a coluna em temperatura ambiente por 15 min; em seguida, lave novamente com 350 µl de tampão de lavagem 1 antes de prosseguir com o protocolo. Realizamos a síntese da primeira cadeia de cDNA a partir de 400 ng de RNA total em reações de 20 µl usando um kit de transcrição reversa Promega GoScript (Promega, catálogo # A5000).
Em seguida, avaliamos os genomas vetoriais e os transcritos virais encontrados em cada amostra usando PCR quantitativo em um Roche Lightcycler II. Aqui, 100 ng de DNA foram usados em uma reação de amplificação de 20 µl usando sondas TaqMan da Thermo Fisher Scientific (sonda EGFP-FAM, ensaio ID Mr04097229_mr, catálogo #4331182; sonda de referência genômica personalizada CN2386-2-VIC, Assay ID ARH6DUK, catálogo # 4448512, projetado para atingir ambos M. mulata e Macaca nemestrina).
Experimentos com macaco verde
Todo o macaco verde (C. sabaeus) os procedimentos foram realizados no Virscio e aprovados pelo IACUC. Todos os macacos foram testados para anticorpos neutralizantes e confirmados como tendo título <1:5. Com aproximadamente 7–8 meses de idade (1.0–1.3 kg), os macacos receberam doses intravenosas (Tabela Suplementar 6). As formulações de dose foram deixadas equilibrar aproximadamente à temperatura ambiente por pelo menos 10 minutos, mas não mais que 60 minutos antes da dosagem. Os volumes de dose IV foram baseados nos pesos corporais do dia 0. Os animais foram sedados com cetamina (8.0 mg kg-1) e xilazina (1.6 mg kg-1). A área de injeção foi raspada e preparada com clorohexidina e isopropanol 70% e esfregada cirurgicamente antes da inserção do cateter intravenoso. A dosagem ocorreu com uma única infusão IV de AAV (7.5 × 1013 ou 7.6 × 1013 peso corporal-1) no dia 0 através da veia safena administrado usando um dispositivo de infusão portátil a uma taxa alvo de 1 ml min-1. O bem-estar geral foi confirmado duas vezes ao dia por observação no lado da gaiola, começando 1 semana antes da administração. No horário programado para a eutanásia, os macacos foram sedados com cetamina (8–10 mg kg-1 intramuscular) e eutanasiado com pentobarbital sódico (100 mg kg-1 IV para efeito). Na perda do reflexo corneano, uma perfusão transcardíaca (ventrículo esquerdo) foi realizada com PBS resfriado usando uma bomba peristáltica a uma taxa de aproximadamente 100 ml min-1 até que o fluido escapado fique claro antes da coleta do tecido. Cubos de tecido foram coletados do hemisfério esquerdo do cérebro e de vários outros órgãos e congelados na fase de vapor de nitrogênio líquido para processamento adicional para biodistribuição. O hemisfério direito do cérebro foi removido e cortado em fatias coronais de ~ 4 mm e pós-fixado intacto com aproximadamente 20 volumes de formalina tamponada neutra a 10% por aproximadamente 24 horas em temperatura ambiente.
O DNA genômico foi extraído do SNC e tecidos periféricos usando o kit de extração Thermo Fisher MagMax DNA Ultra 2.0 (catálogo # A36570). O rendimento do DNA foi avaliado por quantificação fluorométrica com o ensaio Qubit dsDNA. Aproximadamente 20 ng de DNA foram carregados em cada 20 μl de reação e as placas foram executadas no sistema de detecção de PCR em tempo real BioRad CFX Connect (catálogo # 1855201). O ensaio do número de cópias virais foi validado quanto à especificidade pela detecção de um único produto amplificado; sensibilidade, avaliando o limite inferior de detecção como superior a dez cópias por reação; e linearidade, garantindo a curva padrão R2 foi >0.95. As reações foram montadas em FastStart Universal SYBR Green Master (Rox) (catálogo # 4913850001). As sequências dos primers foram ACGACTTCTTCAAGTCCGCC (forward) e TCTTGTAGTTGCCGTCGTCC (reverse). O protocolo de PCR utilizou uma etapa inicial de desnaturação de 95 °C por 180 s, seguida por 40 ciclos de 95 °C por 15 s e 60 °C por 60 s, com uma etapa de imagem após cada ciclo de 60 °C. Uma curva padrão foi gerada com plasmídeo linearizado contendo a sequência modelo GFP presente no vírus de 1 × 108 para 1 × 1010 cópias, diluídas em amostras de DNA de macaco não tratadas, preparadas usando um kit idêntico às amostras deste estudo para controlar os efeitos da matriz. Cópias de DNA viral foram calculadas a partir da curva padrão usando a equação da linha de melhor ajuste. Os valores de multiplicidade de infecção foram calculados com base no peso genômico total medido do DNA da célula hospedeira por reação.
Após a fixação, os tecidos foram colocados em sacarose a 10% > 20% > 30% por 24 h cada a 4 °C e depois embebidos em O.C.T. composto e armazenado a -80 °C até a criossecção. Os blocos de tecido foram levados a -20 °C em um criostato antes de serem seccionados em fatias de 30 μm e montados a seco em lâminas após a criossecção. Após o corte, as lâminas foram deixadas em temperatura ambiente durante a noite para secar. Para auxiliar na quantificação de neurônios, coramos seções com os seguintes anticorpos e concentrações: anti-GFP de coelho (1:100; Millipore-Sigma, cat # AB3080; RRID: AB_91337) e anti-NeuN de camundongo (A60) (1:500; Millipore-Sigma, cat # MAB377; RRID: AB_2298772). Para coloração de anticorpos secundários, foram utilizados os seguintes anticorpos secundários e concentrações: Alexa Fluor 488 anti-coelho de burro (1:500; Thermo Fisher Scientific, cat # A-21206; RRID: AB_2535792) e Alexa Fluor 647 anti-rato de burro (1 :500; Thermo Fisher Scientific, cat # A-31571; RRID: AB_162542). Todos os anticorpos foram diluídos com 1x PBS suplementado com 0.25% de Triton X-100 (PBST) e 5.00% de soro normal de burro. As incubações de anticorpos primários foram deixadas durante a noite à temperatura ambiente. As seções foram então lavadas com PBST. As incubações de anticorpos secundários foram realizadas durante 2 horas à temperatura ambiente. As seções foram lavadas três vezes em PBST. As secções foram incubadas em solução DAPI (1:10,000; Invitrogen, D1306) à temperatura ambiente durante 5 minutos e depois lavadas. As seções foram cobertas com Prolong Diamond Antifade.
Três seções por animal foram coradas e fotografadas. Cada seção foi fotografada em triplicata, com cada região de interesse tendo um total de nove imagens. As regiões de interesse do tecido foram fotografadas com um Keyence BZ-X800 com os seguintes parâmetros de aquisição: GFP (1/500 s), Cy5 (1 s), DAPI (1/12 s), alta resolução Z empilhar em passo de 1.2 µm. As seguintes sub-regiões cerebrais foram visualizadas: córtices frontal, parietal, temporal, occipital, cerebelo, caudado, putâmen e tálamo (núcleos medial, ventral lateral e ventral posterior). Um método semiautomático de contagem de células foi realizado utilizando ImageJ (RRID: SCR_003070) para quantificação. Usando limiares e análise de partículas, quantificamos células positivas para NeuN e positivas para DAPI. Usando o contador de células do ImageJ, contamos manualmente as células positivas para GFP, bem como as células duplamente positivas para GFP e NeuN.
Experimentos iPSC
As culturas neuronais foram produzidas diferenciando e amadurecendo células progenitoras neurais derivadas de iPSC com kits de diferenciação e maturação Stemdiff Forebrain (StemCell # 08600 e # 08605, respectivamente), de acordo com os protocolos do fabricante. Células progenitoras neurais foram produzidas por diferenciação da linha iPSC derivada de fibroblastos do prepúcio: ACS-1019 (ATCC# DYS-0100; RRID: CVCL_X499), com kits de indução neural Stemdiff SMADi (StemCell l#08581), seleção com Stemdiff Neural Rosette Selection Reagente (StemCell l#05832) e expansão em Stemdiff Neural Progenitor Media (StemCell l#05833), conforme protocolos do fabricante. Os neurônios foram amadurecidos por um mínimo de 8 dias antes de serem replantados para transdução.
Culturas neuronais maduras, semeadas com 15,000 células por poço em placas ópticas de 96 poços revestidas com poliornitina e laminina, foram cultivadas por mais 4 dias antes da transdução. Os poços replicados foram transduzidos com vírus diluídos em série em seis ordens de grandeza em meio de maturação de 90% e OptiPRO SFM a 10%. Quatro dias após a transdução, as culturas foram fixadas com 4% de PFA e contrastadas com 1 µg ml-1 Hoechst 33322. A identificação de células transduzidas foi determinada por imagem de 60 campos por poço, usando detecção de fluorescência de dois canais (Hoechst, ex386/em440; eGFP, ex485/em521) em uma plataforma CellInsight CX5 HCS. Células individuais foram identificadas pela detecção Hoechst de seus núcleos e pela aplicação de máscaras de anel com restrição de tamanho e contato a cada célula. A transdução celular foi determinada medindo a fluorescência do eGFP acima de um nível limite dentro de uma máscara de anel individual. Para cada população, a percentagem de células transduzidas foi representada graficamente versus a dose aplicada. Ajustes de curva e CE50 os valores foram determinados com o método de regressão agonista versus resposta (três parâmetros) Prism GraphPad (RRID: SCR_002798). Para relatar eficiências de expressão de eGFP por célula, as intensidades de fluorescência pontual de eGFP foram calculadas em média a partir de cada máscara de anel em um mínimo de 5,000 células por poço. Os ajustes da curva foram obtidos utilizando o Prism GraphPad Biphasic X como método de regressão de concentração.
Estatística e reprodutibilidade
Para as imagens representativas, pelo menos três fatias separadas de cada amostra foram montadas para geração de imagens. Dentro de cada região do cérebro de um único animal, pelo menos três campos de visão diferentes foram obtidos (campo de visão mínimo após o ladrilho, 2.38 mm × 2.38 mm; espessura do corte, 50 µm), equivalendo a nove campos de visão separados em três fatias cerebrais , para garantir consistência entre amostras de imagem.
Resumo de relatórios
Mais informações sobre projeto de pesquisa estão disponíveis no site Resumo do Relatório do Portfólio da Natureza vinculado a este artigo.
- Conteúdo com tecnologia de SEO e distribuição de relações públicas. Seja amplificado hoje.
- PlatoData.Network Gerativa Vertical Ai. Capacite-se. Acesse aqui.
- PlatoAiStream. Inteligência Web3. Conhecimento Amplificado. Acesse aqui.
- PlatãoESG. Automotivo / EVs, Carbono Tecnologia Limpa, Energia, Ambiente, Solar, Gestão de resíduos. Acesse aqui.
- BlockOffsets. Modernizando a Propriedade de Compensação Ambiental. Acesse aqui.
- Fonte: https://www.nature.com/articles/s41565-023-01419-x
- :é
- :onde
- $UP
- 000
- 1
- 10
- 100
- 11
- 12
- 13
- 14
- 15%
- 16
- 17
- 180
- 2%
- 20
- 200
- 2011
- 2015
- 2016
- 2017
- 2018
- 2019
- 2020
- 2021
- 24
- 25
- 27
- 30
- 300
- 31
- 33
- 39
- 40
- 50
- 500
- 60
- 66
- 7
- 70
- 72
- 75
- 8
- 80
- 9
- 95%
- a
- acima
- absoluto
- Acesso
- conformidade
- Segundo
- Alcançar
- aquisição
- em
- ativo
- atividade
- Ad
- adicionar
- adicionado
- Adicional
- Adicionalmente
- administrado
- administração
- adotado
- Adulto
- avançado
- adverso
- Depois de
- novamente
- idade
- AL
- Alexa
- algoritmo
- alinhado
- Todos os Produtos
- permitidas
- juntamente
- tb
- quantidade
- Amplificação
- Amplificado
- amplificando
- an
- análise
- Apresentadora
- e
- animal
- animais
- Anticorpos
- qualquer
- aplicado
- Aplicando
- apropriado
- aprovou
- aproximadamente
- ÁREA
- por aí
- artigo
- artificial
- AS
- montado
- Montagem
- avaliado
- Avaliando
- auxiliar
- At
- Automatizado
- disponível
- média
- longe
- Espinha dorsal
- BANDA
- baseado
- BD
- BE
- passou a ser
- antes
- Começo
- ser
- referência
- MELHOR
- entre
- viés
- obrigatório
- Preto
- Bloquear
- Blocos
- sangue
- corpo
- nascido
- ambos
- BP
- Cérebro
- miolos
- brevemente
- Trazido
- amortecer
- mas a
- by
- gaiolas
- calcular
- calculado
- Califórnia
- CAN
- capturar
- capturados
- Cuidado
- Carga
- CAT
- célula
- Células
- celular
- Centralização de
- central
- cadeia
- chan
- alterar
- mudado
- escolhido
- remover filtragem
- clique
- de perto
- agrupamento
- coleção
- Coluna
- combinado
- combinando
- comitê
- comum
- complementar
- completar
- Efetuado
- compliance
- obedecer
- Compound
- concentração
- condição
- condições
- Confirmar
- CONFIRMADO
- Contato
- conectado
- constante
- construir
- contida
- contém
- ao controle
- controlador
- cópias
- núcleo
- coronal
- Contador
- critérios
- Cultura
- curva
- personalizadas
- Feito sob encomenda
- Cortar
- ciclo
- ciclos
- diariamente
- dados,
- conjuntos de dados
- Davis
- dia
- dias
- profundo
- definido
- entregue
- Entrega
- descrito
- Design
- projetado
- desejado
- detalhes
- Detecção
- Determinar
- determinado
- desvio
- dispositivo
- Dispositivos/Instrumentos
- Diamante
- Diego
- diferenças
- diferente
- diluição
- Ecrã
- exibido
- distância
- distinto
- distribuição
- diversificado
- Diversidade
- dna
- feito
- dosar
- dosagem
- distância
- secar
- duração
- durante
- e
- E & T
- cada
- Mais cedo
- edição
- efeito
- efeitos
- eficiências
- eficiente
- eficientemente
- ou
- elementos
- eliminando
- incorporado
- Inglaterra
- garantir
- assegurando
- estabelecido
- Éter (ETH)
- EU
- avaliadas
- Cada
- exemplo
- exchange
- expansão
- experimentar
- experimentos
- expressão
- extrato
- Extração
- família
- Alimentado
- feminina
- campo
- Campos
- Figo
- Figura
- Arquivos
- filtragem
- final
- Finalmente
- descobertas
- Primeiro nome
- caber
- apropriado
- cinco
- fixado
- Flash
- fluxo
- fluido
- seguido
- seguinte
- segue
- comida
- Escolha
- para rendimento
- para a frente
- encontrado
- quatro
- QUADRO
- Gratuito
- da
- congelado
- funcional
- mais distante
- GAS
- calibre
- Geral
- gerar
- gerado
- geração
- Genética
- genoma
- genómica
- dado
- gráfico
- maior
- Verde
- Grupo
- Do grupo
- orientações
- Metade
- Manipulação
- Ter
- ter
- cabeça
- Saúde
- SUA PARTICIPAÇÃO FAZ A DIFERENÇA
- Alta
- de alta resolução
- Buracos
- homogeneizado
- hospedeiro
- HTTPS
- humano
- ICE
- ID
- idêntico
- identificação
- identificado
- if
- ii
- iii
- imagens
- Imagiologia
- imerso
- imersão
- melhorar
- in
- incluído
- incubado
- independentemente
- índice
- indicado
- Índices
- Individual
- Individualmente
- indução
- infection
- INFORMAÇÕES
- infundido
- infusão
- do estado inicial,
- inicialmente
- em vez disso
- Instituto
- DOCUMENTOS
- instrumentos
- interesse
- Interface
- para dentro
- Intravenosa
- introduzido
- envolvido
- isolado
- Jackson
- de emergência
- laboratório
- laboratório
- Laboratório
- Maior
- ld
- mínimo
- esquerda
- menos
- Nível
- bibliotecas
- Biblioteca
- vida
- LIMITE
- Line
- LINK
- ligado
- Líquido
- Lista
- Fígado
- local
- longo prazo
- fora
- diminuir
- baixo
- moldadas
- manutenção
- manualmente
- mapeamento
- máscara
- Máscaras
- dominar
- Match
- correspondente
- material
- Matriz
- a medida
- medido
- medição
- Mídia
- média
- mental
- A saúde mental
- método
- métodos
- métrico
- camundongos
- Microscópio
- minutos
- mínimo
- minutos
- misturar
- mistura
- ML
- modificada
- monitorados
- Mês
- mês
- mais
- mRNA
- MSCI
- nomeadamente
- nanotecnologia
- Nacional
- Natureza
- Perto
- necessário
- rede
- redes
- Neural
- neuronal
- Neurônios
- Novo
- Próximo
- NIH
- não
- nós
- normal
- número
- objetivo
- obtido
- ocorreu
- of
- Velho
- on
- ONE
- só
- ideal
- otimizado
- or
- ordens
- Outros
- A Nossa
- Acima de
- durante a noite
- Oxford
- Oxygen
- acondicionamento
- emparelhado
- parâmetros
- parte
- partícula
- PBS
- PCR
- Pico
- Penn
- Pennsylvania
- para
- percentagem
- perfeita
- realizada
- periférico
- fase
- oleoduto
- Passo
- Lugar
- planejado
- Plasma
- plataforma
- platão
- Inteligência de Dados Platão
- PlatãoData
- mais
- piscina
- população
- pasta
- Precisão
- preparação
- preparado
- presente
- apresentado
- evitar
- anteriormente
- primário
- primeiro
- sonda
- procedimentos
- procedimentos
- processo
- processado
- em processamento
- produzir
- Produzido
- Produto
- Produção
- Produtos
- progenitor
- Agenda
- protetor
- Proteína
- Proteínas
- protocolo
- protocolos
- fornece
- publicado
- bomba
- comprado
- Python
- qualidade
- quantitativo
- Qubit
- Coelho
- angariado
- Taxa
- relação
- alcançar
- reação
- reações
- Leia
- em tempo real
- recebido
- Recomenda
- recuperação
- região
- regiões
- regressão
- reguladores
- relativo
- remover
- Removido
- replica
- Denunciar
- Informou
- Relatórios
- representante
- representado
- requeridos
- pesquisa
- respectivamente
- resposta
- respostas
- restrição
- resultando
- reverso
- certo
- Anel
- Subir
- ascensão
- RNA
- uma conta de despesas robusta
- rocha
- Quarto
- Rota
- Execute
- é executado
- s
- mesmo
- San
- San Diego
- arenoso
- Escala
- programado
- SCI
- científico
- Ponto
- pontuações
- peneiramento
- Scripts
- secundário
- Seção
- seções
- Vejo
- segmentação
- selecionado
- doadores,
- seletivo
- Sensibilidade
- enviei
- separado
- Seqüência
- sequenciamento
- Sérum
- conjunto
- Configurações
- vários
- enviado
- mostrar
- Sides
- Vista
- Signal
- semelhante
- solteiro
- sentar-se
- SIX
- tamanhos
- Pele
- Fatia
- slide
- Slides
- Lentamente
- pequeno
- sódio
- solução
- Parecer
- especificamente
- especificidade
- Spot
- pilha
- Pilhas
- padrão
- Passo
- Passos
- armazenamento
- armazenadas
- Tanga
- Estudo
- subseqüente
- adequado
- topo
- superfície
- sintético
- .
- sistêmico
- sistemas
- mesa
- tomado
- tocando
- Target
- visadas
- alvejando
- Tecnologias
- Tecnologia
- modelo
- dez
- terminal
- ensaio
- do que
- que
- A
- A área
- deles
- Eles
- então
- Lá.
- assim sendo
- Este
- deles
- isto
- três
- limiar
- Através da
- todo
- tempo
- vezes
- tecidos
- para
- levou
- Total
- traçar
- Traçado
- transferência
- transferido
- tratado
- Tritão
- Twice
- torção
- dois
- tipos
- Ultra
- para
- único
- Universal
- universidade
- Universidade da Califórnia
- Universidade da Pensilvânia
- até
- usar
- usava
- utilização
- utilizar
- Utilizando
- validado
- Valores
- Variante
- vário
- versão
- Contra
- via
- Ver
- viral
- vírus
- vírus
- vivo
- volumes
- foi
- Washington
- Água
- we
- semana
- semanas
- peso
- BEM
- Wells
- foram
- qual
- inteiro
- generalizada
- de
- dentro
- mundo
- X
- anos
- Produção
- rendimentos
- Zen
- zefirnet