Agencja ds. Żywności i Leków (FDA) odgrywa kluczową rolę w ochronie zdrowia publicznego poprzez regulacje dotyczące wyrobów medycznych, zapewniając, że są one bezpieczne i skuteczne w przypadku ich zamierzonego zastosowania. W dzisiejszych czasach wzrost liczby podłączonych urządzeń sprawił, że cyberbezpieczeństwo znalazło się na pierwszym miejscu wśród obaw FDA; w tym kontekście istotne jest określenie szczegółowych wymagań dotyczących definicji modelu zagrożeń cyberbezpieczeństwa. W miarę jak wyroby medyczne stają się coraz bardziej zintegrowane z technologią, potrzeba solidnych środków cyberbezpieczeństwa w celu ochrony informacji o pacjentach i zapewnienia funkcjonalności tych urządzeń jest bardziej krytyczna niż kiedykolwiek. Zgodność z przepisami FDA jest niezbędna nie tylko dla zgodności producentów, ale także dla bezpieczeństwa i zaufania pacjentów i użytkowników.
Kilkakrotnie omawialiśmy cyfrowe urządzenia medyczne i powiązane wymagania na stronach internetowych QualityMedDev, poruszając różne tematy, takie jak Sztuczna inteligencja w urządzeniach medycznych, cyfrowe produkty zdrowotne, Podejście TGA do regulacji cyfrowych urządzeń medycznych. Jednocześnie FDA opublikowała różne wytyczne w odniesieniu do cyberbezpieczeństwa, zarówno w odniesieniu do wymagania przed wprowadzeniem na rynek i wymagania po wprowadzeniu na rynek.
W ramach nadzoru regulacyjnego FDA ustanowiła wymogi dotyczące cyberbezpieczeństwa w celu ochrony pacjentów i zapewnienia integralności wyrobów medycznych. Normy te dotyczą całego cyklu życia urządzenia, od początkowego projektu i rozwoju po wdrożenie i konserwację. Ponieważ urządzenia medyczne stają się coraz inteligentniejsze i lepiej połączone, są one coraz bardziej podatne na zagrożenia cybernetyczne. Integracja praktyk w zakresie cyberbezpieczeństwa na etapie opracowywania wyrobów medycznych jest kluczowym krokiem w kierunku ograniczenia ryzyka stwarzanego przez cyberataki.
Kluczowe elementy ram cyberbezpieczeństwa FDA
Aby zapewnić integralność cyberbezpieczeństwa wyrobów medycznych, FDA określiła wymagania przed wprowadzeniem na rynek, które obejmują potrzebę uwzględnienia przez producentów modelu zagrożeń cyberbezpieczeństwa i mechanizmów kontroli bezpieczeństwa już na najwcześniejszych etapach koncepcji urządzenia.
Producenci wyrobów ustanawiają systemy jakości i przestrzegają ich, aby zapewnić stałą zgodność swoich produktów z obowiązującymi wymaganiami i specyfikacjami. Wymagania dotyczące tych systemów jakości można znaleźć w przepisach dotyczących systemu jakości (QS) w 21 CFR część 820, jeśli urządzenie jest sprzedawane w Stanach Zjednoczonych, lub w innych przepisach dla innych krajów (np. UE MDR 2017/745 dla Unii Europejskiej).
W zależności od charakteru urządzenia wymagania QS mogą mieć zastosowanie na etapie przed wprowadzeniem na rynek, na etapie po wprowadzeniu do obrotu lub na obu etapach. W kontekście fazy przed wprowadzeniem na rynek wykazanie wystarczającej pewności co do bezpieczeństwa i skuteczności niektórych urządzeń stwarzających zagrożenia dla cyberbezpieczeństwa może obejmować uwzględnienie wyników dokumentacji związanej z rozporządzeniem w sprawie jakości w ramach składania przed wprowadzeniem na rynek. Na przykład ISO 13485:2016 i 21 CFR 820 nakładają na producentów wszystkich klas urządzeń zautomatyzowanych za pomocą oprogramowania obowiązek ustanowienia procedur kontroli projektu urządzenia, zapewniających zgodność z określonymi wymaganiami projektowymi (zwanych „kontrolami projektu”). W ramach kontroli projektu producenci są zobowiązani do „ustanowienia i utrzymywania procedur walidacji projektu urządzenia”, co obejmuje, w stosownych przypadkach, „walidację oprogramowania i analizę ryzyka”. W ramach obowiązkowej walidacji oprogramowania i analizy ryzyka producenci oprogramowania mogą być zmuszeni do wdrożenia procesów zarządzania ryzykiem cybernetycznym i walidacji, jeśli ma to zastosowanie.
Walidacja oprogramowania i zarządzanie ryzykiem to kluczowe elementy analiz cyberbezpieczeństwa, określające, czy urządzenie zapewnia wystarczającą pewność bezpieczeństwa i skuteczności. FDA nakazuje producentom włączenie procesów rozwoju, które uwzględniają i eliminują ryzyko oprogramowania na wszystkich etapach projektowania i rozwoju, w ramach kontroli projektu. Procesy te powinny uwzględniać kwestie cyberbezpieczeństwa. Obejmuje to identyfikację zagrożeń bezpieczeństwa, ustalenie wymagań projektowych w celu kontrolowania tych zagrożeń oraz przedstawienie dowodów, że kontrole działają zgodnie z przeznaczeniem i są skuteczne w wyznaczonym środowisku urządzenia, zapewniając wystarczające środki bezpieczeństwa.
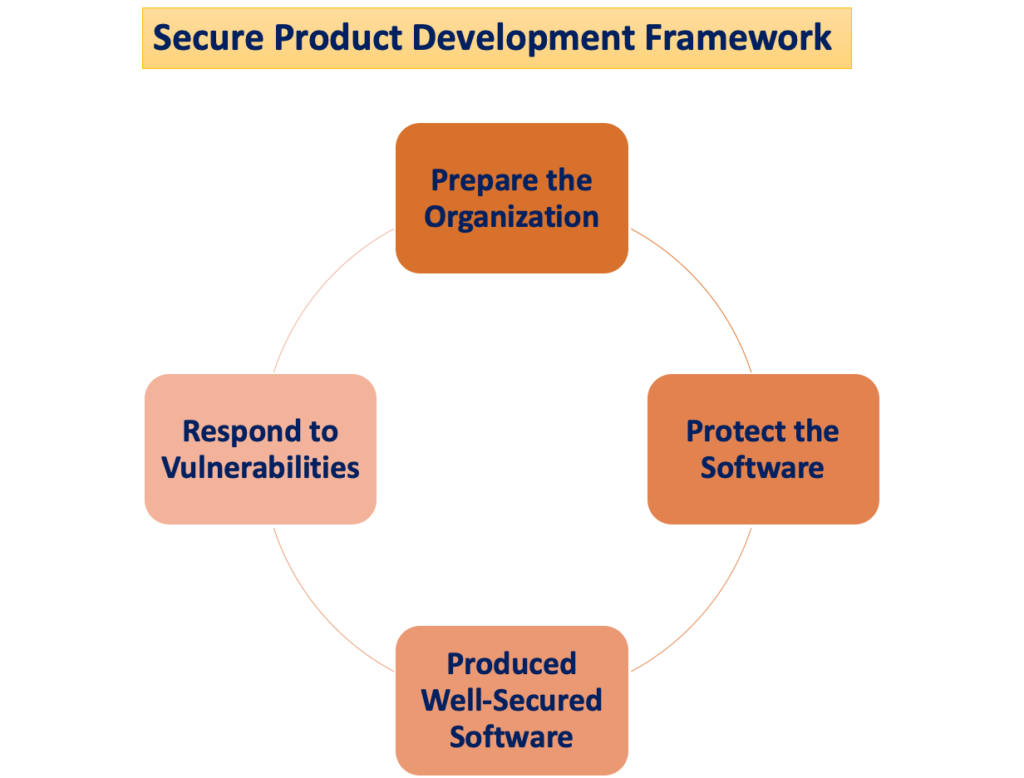
"Bezpieczne ramy rozwoju produktu"
Potencjał wyrządzenia szkody pacjentowi pojawia się, gdy zagrożenia cyberbezpieczeństwa wykorzystują luki w systemie, a łatwość, z jaką te zagrożenia mogą zagrozić bezpieczeństwu i skuteczności wyrobu medycznego, zwiększa się z czasem wraz z liczbą zidentyfikowanych luk. Bezpieczne ramy rozwoju produktu (SPDF) składają się z procesów mających na celu identyfikację i zmniejszenie ilości i wagi luk w zabezpieczeniach produktów. Obejmując wszystkie etapy cyklu życia produktu – projektowanie, rozwój, wydawanie, wsparcie i likwidację – SPDF jest integralną częścią.
Włączenie procesów SPDF na etapie projektowania urządzenia może zapobiec konieczności ponownego projektowania podczas integrowania funkcji opartych na łączności po wprowadzeniu na rynek lub usuwania luk w zabezpieczeniach, które stwarzają niekontrolowane ryzyko. Możliwa integracja z istniejącymi procesami rozwoju produktów i oprogramowania, zarządzania ryzykiem i szerszym systemem jakości zwiększa wszechstronność SPDF.
Aby zapewnić zgodność z przepisami dotyczącymi systemu jakości (QS), zaleca się stosowanie SPDF. FDA zachęca producentów do stosowania SPDF ze względu na korzyści związane z spełnianiem zarówno przepisów dotyczących QS, jak i wymogów cyberbezpieczeństwa. Uznaje się jednak, że podejścia alternatywne mogą również spełniać wymagania przepisów dotyczących jakości.
Zarządzanie ryzykami cyberbezpieczeństwa
Podstawowym celem stosowania SPDF (Secure Product Development Framework) jest tworzenie i utrzymywanie urządzeń, które są zarówno bezpieczne, jak i skuteczne. Z punktu widzenia bezpieczeństwa urządzenia te charakteryzują się również niezawodnością i odpornością. Producenci i/lub użytkownicy (np. pacjenci, placówki opieki zdrowotnej) mogą następnie zarządzać tymi urządzeniami, w tym instalacją, konfiguracją, aktualizacjami i przeglądaniem dzienników urządzenia, poprzez projekt urządzenia i powiązane etykietowanie.
Placówki opieki zdrowotnej mają możliwość zarządzania tymi urządzeniami w ramach własnych ram zarządzania ryzykiem cyberbezpieczeństwa, takich jak powszechnie uznany Narodowy Instytut Standardów i Technologii (NIST) Ramy Poprawy Cyberbezpieczeństwa Infrastruktury Krytycznej, powszechnie zwane NIST Cyberbezpieczeństwo Framework lub NIST CSF.
FDA zaleca, aby producenci wdrożyli procesy projektowania urządzeń, takie jak te określone w przepisach dotyczących systemu jakości (QS), aby zwiększyć bezpieczeństwo rozwoju i konserwacji produktów. Zachowując elastyczność dla producentów, mogą oni również zbadać alternatywne ramy zgodne z rozporządzeniem w sprawie jakości i przestrzegać zaleceń FDA dotyczących wdrażania SPDF. Przykłady obejmują ramy dotyczące konkretnych wyrobów medycznych we wspólnym planie bezpieczeństwa wyrobów medycznych i zdrowia IT (JSP) 30 oraz IEC 81001-5-1. Ramy z innych sektorów, takie jak ANSI/ISA 62443-4-1 Bezpieczeństwo systemów automatyki przemysłowej i sterowania Część 4-1: Wymagania dotyczące cyklu życia rozwoju bezpieczeństwa produktu, mogą również spełniać przepisy dotyczące QS.
W kolejnych sekcjach tego artykułu przedstawiono zalecenia dotyczące stosowania procesów SPDF, zgodnie z ogólną oceną FDA, które podkreślają kluczowe kwestie dotyczące opracowywania urządzeń, które są bezpieczne i skuteczne. Procesy te uzupełniają regulacje dotyczące QS, a FDA sugeruje producentom dołączenie odpowiedniej dokumentacji do przeglądu w zgłoszeniach przed wprowadzeniem na rynek.
Model zagrożeń cyberbezpieczeństwa
Modelowanie zagrożeń obejmuje systematyczny proces identyfikacji celów bezpieczeństwa, zagrożeń i słabych punktów w całym systemie wyrobów medycznych. Następnie wiąże się to ze zdefiniowaniem środków zaradczych mających na celu zapobieganie, łagodzenie, monitorowanie lub reagowanie na skutki zagrożeń w całym cyklu życia systemu wyrobu medycznego. Odpowiednio i kompleksowo zastosowany stanowi podstawę optymalizacji bezpieczeństwa komponentów systemu, produktów, sieci, aplikacji i połączeń.
Jeśli chodzi o zarządzanie ryzykiem bezpieczeństwa oraz w celu określenia odpowiednich zagrożeń bezpieczeństwa i kontroli systemu wyrobów medycznych, FDA zaleca wdrożenie modelowania zagrożeń w celu informowania i wspierania działań związanych z analizą ryzyka. W kontekście oceny ryzyka FDA sugeruje integrację modelowania zagrożeń w całym procesie projektowania, obejmującym wszystkie elementy systemu wyrobu medycznego.
Kluczowe aspekty modelu zagrożeń powinny obejmować identyfikację ryzyk i środków łagodzących, informując o ryzyku zarówno przed, jak i po łagodzeniu, uwzględnianych w ocenie ryzyka cyberbezpieczeństwa. Ponadto model powinien formułować założenia dotyczące systemu wyrobów medycznych lub środowiska użytkowania, takie jak założenie, że sieci szpitalne są z natury wrogie. To założenie skłania producentów do rozważenia scenariuszy, w których przeciwnik kontroluje sieć i może zmieniać, odrzucać i odtwarzać pakiety. Co więcej, model zagrożenia powinien uwzględniać ryzyka cyberbezpieczeństwa wprowadzone w łańcuchu dostaw, produkcji, wdrażaniu, współpracy z innymi urządzeniami, czynnościach konserwacyjnych/aktualizacyjnych i likwidacyjnych, które mogą zostać przeoczone w tradycyjnym procesie oceny ryzyka bezpieczeństwa.
W przypadku zgłoszeń przed wprowadzeniem na rynek FDA zaleca dołączenie dokumentacji modelowania zagrożeń w celu zaprezentowania analizy systemu wyrobów medycznych i identyfikacji potencjalnych zagrożeń bezpieczeństwa, które mogą mieć wpływ na bezpieczeństwo i skuteczność. Producenci mają swobodę wyboru spośród różnych metodologii lub kombinacji metod modelowania zagrożeń, a uzasadnienie wybranych metod należy dołączyć do dokumentacji.
Zaleca się przeprowadzanie działań związanych z modelowaniem zagrożeń podczas przeglądów projektu, a dokumentacja powinna dostarczać wystarczających informacji, aby FDA mogła ocenić i dokonać przeglądu zabezpieczeń zintegrowanych z urządzeniem. Ta holistyczna ocena powinna uwzględniać zarówno urządzenie, jak i szerszy system, w którym pracuje, podkreślając bezpieczeństwo i skuteczność urządzenia.
Główne elementy modeli zagrożeń cyberbezpieczeństwa można podsumować w następujący sposób:
Identyfikacja potencjalnych zagrożeń
Opracowanie modelu zagrożeń stanowi podstawowy krok w procesie cyberbezpieczeństwa, umożliwiając producentom identyfikację i zrozumienie potencjalnych zagrożeń cyberbezpieczeństwa charakterystycznych dla ich wyrobów medycznych. Opracowanie modelu zagrożeń stanowi podstawowy krok w procesie cyberbezpieczeństwa, umożliwiając producentom identyfikację i zrozumienie potencjalnych zagrożeń cyberbezpieczeństwa charakterystycznych dla ich wyrobów medycznych. Opracowanie modelu zagrożeń stanowi podstawowy krok w procesie cyberbezpieczeństwa, umożliwiając producentom identyfikację i zrozumienie potencjalnych zagrożeń cyberbezpieczeństwa charakterystycznych dla ich wyrobów medycznych.
Ocena i zarządzanie ryzykiem
Ocena i zarządzanie ryzykiem polega na ocenie zidentyfikowanych zagrożeń w celu ustalenia ich potencjalnego wpływu i opracowaniu odpowiednich strategii w celu skutecznego ograniczenia tych ryzyk.
Wdrożenie kontroli bezpieczeństwa
Kontrole bezpieczeństwa to zabezpieczenia lub środki zaradcze wdrożone w celu ochrony poufności, integralności i dostępności wyrobu medycznego przed zidentyfikowanymi zagrożeniami.
Przyszłe trendy w wytycznych FDA dotyczących cyberbezpieczeństwa
W miarę ewolucji zagrożeń i technologii zmieniają się także wytyczne FDA dotyczące cyberbezpieczeństwa. Producenci muszą być na bieżąco informowani i sprawnie reagować na te zmiany. Producenci powinni przewidywać aktualizacje przepisów, śledząc trendy branżowe i zachowując proaktywne podejście do cyberbezpieczeństwa.
Przygotowanie na nieprzewidziane wyzwania ma kluczowe znaczenie; ustanowienie solidnych protokołów bezpieczeństwa i przyszłościowych strategii umożliwi producentom skuteczne reagowanie na przyszły stan przepisów dotyczących cyberbezpieczeństwa.
Cyberbezpieczeństwo to aspekt regulacji dotyczących wyrobów medycznych, którego nie można przecenić — jest on tak samo nieodłączny od bezpieczeństwa urządzenia, jak każda funkcja mechaniczna lub elektryczna. FDA uznała to i wdrażając kompleksowe ramy cyberbezpieczeństwa, stara się dotrzymać kroku innowacjom, jednocześnie chroniąc zdrowie publiczne. Producenci, pracownicy służby zdrowia i pacjenci muszą współpracować, aby przestrzegać tych standardów i zapewnić ciągłą niezawodność i bezpieczeństwo wyrobów medycznych w obliczu zagrożeń cybernetycznych.
Zapisz się do newslettera QualityMedDev
QualityMedDev to platforma internetowa skoncentrowana na zagadnieniach dotyczących jakości i przepisów w branży urządzeń medycznych; Śledź nas na LinkedIn i Twitter aby być na bieżąco z najważniejszymi wiadomościami w dziedzinie przepisów.
QualityMedDev to jedna z największych platform internetowych wspierających biznes urządzeń medycznych w zakresie zgodności z przepisami. Zapewniamy usługi doradztwa regulacyjnego, w szerokim zakresie tematów, od UE MDR i IVDR do ISO 13485, w tym zarządzanie ryzykiem, biokompatybilność, weryfikację i walidację użyteczności i oprogramowania oraz ogólnie wsparcie w przygotowaniu dokumentacji technicznej dla MDR.
Nasza siostrzana platforma Akademia JakościMedDev zapewnia możliwość udziału w kursach szkoleniowych online i we własnym tempie, skoncentrowanych na zagadnieniach zgodności z przepisami dotyczącymi urządzeń medycznych. Te kursy szkoleniowe, opracowane we współpracy z wysoko wykwalifikowanymi specjalistami z branży urządzeń medycznych, pozwalają wykładniczo zwiększyć Twoje kompetencje w szerokim zakresie zagadnień dotyczących jakości i przepisów dotyczących działalności biznesowej związanej z urządzeniami medycznymi.

Nie wahaj się zapisać do naszego Newslettera!
- Dystrybucja treści i PR oparta na SEO. Uzyskaj wzmocnienie już dziś.
- PlatoData.Network Pionowe generatywne AI. Wzmocnij się. Dostęp tutaj.
- PlatoAiStream. Inteligencja Web3. Wiedza wzmocniona. Dostęp tutaj.
- PlatonESG. Węgiel Czysta technologia, Energia, Środowisko, Słoneczny, Gospodarowanie odpadami. Dostęp tutaj.
- Platon Zdrowie. Inteligencja w zakresie biotechnologii i badań klinicznych. Dostęp tutaj.
- Źródło: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- :ma
- :Jest
- :nie
- :Gdzie
- $W GÓRĘ
- 2016
- 30
- 350
- 820
- 9
- a
- O nas
- Akademia
- przyznał
- w poprzek
- zajęcia
- do tego
- adres
- adresowanie
- Dodaje
- przylegać
- administracja
- rozmyślny
- Zwolennicy
- zwinny
- wymierzony
- Cele
- wyrównać
- Wszystkie kategorie
- pozwala
- również
- alternatywny
- an
- analizuje
- analiza
- i
- przewidywać
- każdy
- odpowiedni
- aplikacje
- stosowany
- Aplikuj
- podejście
- awanse
- właściwy
- odpowiednio
- SĄ
- artykuł
- AS
- aspekt
- aspekty
- oszacować
- oszacowanie
- powiązany
- założenie
- Założenia
- zapewnienie
- At
- Ataki
- zautomatyzowane
- Automatyzacja
- dostępność
- BE
- stają się
- staje
- być
- Korzyści
- grzbiet
- obie
- szeroki
- szerszy
- przyniósł
- biznes
- operacje biznesowe
- ale
- by
- CAN
- nie może
- zdolny
- zdobyć
- pewien
- łańcuch
- wyzwania
- Zmiany
- Dodaj
- wybrany
- Klasy
- współpracować
- współpraca
- COM
- kombinacje
- powszechnie
- Komplement
- spełnienie
- składniki
- wszechstronny
- kompromis
- konstrukcja
- Obawy
- poufność
- systemu
- połączony
- Podłączonych urządzeń
- połączenia
- Rozważać
- Rozważania
- za
- zgodny
- składa się
- stanowić
- consulting
- współczesny
- kontekst
- nadal
- kontrola
- kontrolowania
- kontroli
- Odpowiedni
- mógłby
- kraje
- kursy
- pokrycie
- Stwórz
- krytyczny
- Infrastruktura krytyczna
- istotny
- cyber
- Ataki komputerowe
- Bezpieczeństwo cybernetyczne
- Data
- definiowanie
- definicja
- demonstrowanie
- Wdrożenie
- Wnętrze
- proces projektowania
- wyznaczony
- określaniu
- rozwinięty
- rozwijanie
- oprogramowania
- urządzenie
- urządzenia
- różne
- cyfrowy
- zmniejsza się
- Omawiając
- dokumentacja
- Rzut
- lek
- podczas
- e
- najwcześniej
- zarabiać
- łatwość
- Efektywne
- faktycznie
- skuteczność
- bądź
- Elementy
- objąć
- podkreślając
- zatrudniający
- umożliwiając
- objąć
- obejmuje
- obejmujący
- zachęca
- zapewnić
- zapewnienie
- Środowisko
- niezbędny
- zapewniają
- ustanowiony
- ustanowienie
- Eter (ETH)
- EU
- europejski
- Unia Europejska
- oceny
- ewaluację
- EVER
- dowód
- ewoluuje
- przykłady
- Przede wszystkim system został opracowany
- Wykorzystać
- odkryj
- wykładniczo
- Twarz
- udogodnienia
- FDA
- wykonalny
- Cecha
- Korzyści
- pole
- Elastyczność
- koncentruje
- obserwuj
- następujący
- następujący sposób
- jedzenie
- Food and Drug Administration
- Food and Drug Administration (FDA)
- W razie zamówieenia projektu
- czoło
- przyszłościowe
- znaleziono
- Fundacja
- Podstawowy
- Framework
- Ramy
- od
- pełny
- Funkcjonalność
- Ponadto
- przyszłość
- Ogólne
- ogólnie
- wytyczne
- zaszkodzić
- Have
- Zdrowie
- opieki zdrowotnej
- Wysoki
- Atrakcja
- wysoko
- holistyczne
- Szpital
- Jednak
- HTML
- HTTPS
- Identyfikacja
- zidentyfikowane
- zidentyfikować
- identyfikacja
- if
- Rezultat
- Oddziaływania
- realizacja
- realizowane
- wykonawczych
- ważny
- poprawy
- in
- zawierać
- obejmuje
- Włącznie z
- włączać
- włączenie
- Zwiększać
- Zwiększenia
- coraz bardziej
- przemysłowy
- automatyka przemysłowa
- przemysł
- informować
- Informacja
- poinformowany
- Infrastruktura
- właściwie
- początkowy
- Innowacja
- instalacja
- przykład
- Instytut
- integralny
- zintegrowany
- Integracja
- integracja
- integralność
- zamierzony
- najnowszych
- wewnętrzny
- wprowadzono
- angażować
- dotyczy
- ISO
- IT
- JEGO
- połączenie
- Trzymać
- Klawisz
- etykietowanie
- największym
- koło życia
- wifecycwe
- MailChimp
- Główny
- utrzymać
- utrzymanie
- konserwacja
- zarządzanie
- i konserwacjami
- mandaty
- Producenci
- produkcja
- Maksymalna szerokość
- Może..
- MDR
- środków
- mechaniczny
- medyczny
- Urządzenie medyczne
- urządzenia medyczne
- Poznaj nasz
- Spotkanie
- metodologie
- metody
- może
- Złagodzić
- łagodzenie
- łagodzenie ryzyka
- model
- modelowanie
- modele
- monitor
- jeszcze
- większość
- musi
- narodowy
- Natura
- Nawigacja
- konieczność
- Potrzebować
- sieć
- sieci
- aktualności
- nist
- numer
- cel
- Cele
- of
- on
- ONE
- Online
- tylko
- działać
- działa
- operacje
- optymalizacji
- Option
- or
- Inne
- ludzkiej,
- opisane
- Wyjścia
- koniec
- Przeoczenie
- własny
- Pokój
- Pakiety
- część
- pacjent
- pacjenci
- spostrzegany
- perspektywa
- faza
- kluczowy
- krok po kroku
- Platforma
- plato
- Analiza danych Platona
- PlatoDane
- odgrywa
- wtyczka
- stwarzane
- position
- możliwość
- Wiadomości
- potencjał
- praktyki
- przygotowanie
- zapobiec
- pierwotny
- Proaktywne
- procedury
- wygląda tak
- procesów
- Produkt
- rozwój produktów
- Produkty
- specjalistów
- monity
- chronić
- ochrony
- protokoły
- zapewniać
- pod warunkiem,
- zapewnia
- że
- publiczny
- zdrowie publiczne
- opublikowany
- jakość
- ilość
- zasięg
- racjonalny
- rozsądny
- uznane
- zalecenia
- Zalecana
- zaleca
- , o którym mowa
- Regulacja
- regulamin
- regulacyjne
- Zgodność z przepisami
- związane z
- relacja
- zwolnić
- niezawodność
- pozostawać
- wymagany
- wymagania
- sprężystość
- Odpowiadać
- przeglądu
- Recenzje
- Rosnąć
- Ryzyko
- ocena ryzyka
- Zarządzanie ryzykiem
- ryzyko
- krzepki
- Rola
- "bezpiecznym"
- zabezpieczenie
- zabezpieczenia
- Bezpieczeństwo
- taki sam
- scenariusze
- działy
- sektor
- Sektory
- bezpieczne
- bezpieczeństwo
- Środki bezpieczeństwa
- zarządzanie ryzykiem bezpieczeństwa
- zagrożenia bezpieczeństwa
- służy
- kilka
- surowość
- powinien
- prezentacja
- siostra
- wykwalifikowany
- mądrzejszy
- So
- Tworzenie
- rozwoju oprogramowania
- sprzedany
- specyficzny
- Specyfikacje
- określony
- STAGE
- etapy
- standardy
- Stan
- Zjednoczone
- pobyt
- przebywający
- Ewolucja krok po kroku
- strategie
- uległość
- Zgłoszenia
- subskrybuj
- Następnie
- taki
- wystarczający
- Wskazuje
- odpowiedni
- Dostawa
- łańcuch dostaw
- wsparcie
- Wspierający
- system
- systemy
- Techniczny
- Technologia
- niż
- że
- Połączenia
- Przyszłość
- ich
- następnie
- Te
- one
- to
- tych
- groźba
- zagrożenia
- Przez
- poprzez
- czas
- czasy
- do
- także
- tematy
- tradycyjny
- Trening
- Trendy
- Zaufaj
- solidność
- zrozumieć
- nieprzewidziany
- unia
- Zjednoczony
- United States
- Nowości
- Utrzymuj
- URL
- us
- użyteczność
- Stosowanie
- posługiwać się
- Użytkownicy
- Wykorzystując
- sprawdzanie poprawności
- uprawomocnienie
- różnorodny
- Weryfikacja
- wszechstronność
- Luki w zabezpieczeniach
- Wrażliwy
- we
- strony internetowe
- jeśli chodzi o komunikację i motywację
- czy
- który
- Podczas
- szeroko
- będzie
- w
- w ciągu
- WordPress
- Wtyczka WordPress
- ty
- Twój
- zefirnet





