La Food and Drug Administration (FDA) svolge un ruolo fondamentale nella salvaguardia della salute pubblica attraverso la regolamentazione dei dispositivi medici, garantendo che siano sicuri ed efficaci per l’uso previsto. Nei tempi contemporanei, l’aumento dei dispositivi connessi ha portato la sicurezza informatica in prima linea tra le preoccupazioni della FDA; in questo contesto, è essenziale definire requisiti specifici per la definizione del modello di minaccia alla sicurezza informatica. Man mano che i dispositivi medici diventano sempre più integrati con la tecnologia, la necessità di solide misure di sicurezza informatica per proteggere le informazioni dei pazienti e garantire la funzionalità di questi dispositivi è più critica che mai. Il rispetto delle normative FDA non è essenziale solo per la conformità dei produttori, ma anche per la sicurezza e la fiducia di pazienti e utenti.
Abbiamo discusso più volte dei dispositivi medici digitali e dei relativi requisiti all'interno dei siti Web QualityMedDev, coprendo diversi argomenti come L’intelligenza artificiale nei dispositivi medici, prodotti sanitari digitalie Approccio TGA per la regolamentazione dei dispositivi medici digitali. Allo stesso tempo, la FDA ha pubblicato diverse linee guida in relazione alla sicurezza informatica, sia in relazione a requisiti pre-mercato ed requisiti post-commercializzazione.
La FDA ha stabilito requisiti di sicurezza informatica come parte della supervisione normativa per proteggere i pazienti e garantire l’integrità dei dispositivi medici. Questi standard si applicano all'intero ciclo di vita di un dispositivo, dalla progettazione e sviluppo iniziali alla distribuzione e manutenzione. Poiché i dispositivi medici diventano sempre più intelligenti e connessi, sono sempre più vulnerabili alle minacce informatiche. L’integrazione delle pratiche di cybersecurity durante la fase di sviluppo dei dispositivi medici è un passo cruciale per mitigare i rischi posti dagli attacchi informatici.
Componenti chiave del quadro di sicurezza informatica della FDA
Per garantire l’integrità della sicurezza informatica dei dispositivi medici, la FDA ha delineato i requisiti pre-commercializzazione che includono la necessità per i produttori di incorporare modelli di minaccia alla sicurezza informatica e controlli di sicurezza fin dalle prime fasi di concezione del dispositivo.
I produttori di dispositivi devono istituire e aderire a sistemi di qualità per garantire la conformità coerente ai requisiti e alle specifiche applicabili per i loro prodotti. I requisiti per questi sistemi di qualità possono essere trovati nel regolamento Sistema Qualità (QS) in 21 CFR Parte 820 se il dispositivo è venduto negli Stati Uniti, o in altri regolamenti per altri paesi (ad esempio EU MDR 2017/745 per l'Unione Europea).
A seconda della natura del dispositivo, i requisiti QS possono essere pertinenti durante la fase pre-commercializzazione, la fase post-commercializzazione o entrambe. Nel contesto della fase pre-commercializzazione, dimostrare una ragionevole garanzia di sicurezza ed efficacia per determinati dispositivi con rischi per la sicurezza informatica può comportare l'inclusione della documentazione relativa al regolamento QS come parte della presentazione pre-commercializzazione. Ad esempio, la norma ISO 13485:2016 e 21 CFR 820 impone che i produttori di tutte le classi di dispositivi automatizzati con software stabiliscano procedure per controllare la progettazione del dispositivo, garantendo la conformità ai requisiti di progettazione specificati (denominati "controlli di progettazione"). Nell'ambito dei controlli di progettazione, i produttori sono tenuti a "stabilire e mantenere procedure per la convalida della progettazione del dispositivo", che comprendono "la convalida del software e l'analisi dei rischi, ove appropriato". Nell’ambito della convalida obbligatoria del software e dell’analisi dei rischi, i produttori di dispositivi software potrebbero dover istituire processi di gestione e convalida dei rischi di sicurezza informatica, ove applicabile.
La convalida del software e la gestione del rischio costituiscono elementi cruciali delle analisi di sicurezza informatica, determinando se un dispositivo fornisce una ragionevole garanzia di sicurezza ed efficacia. La FDA impone ai produttori di incorporare processi di sviluppo che considerino e affrontino i rischi del software durante le fasi di progettazione e sviluppo come parte dei controlli di progettazione. Questi processi dovrebbero comprendere considerazioni sulla sicurezza informatica. Ciò include l'identificazione dei rischi per la sicurezza, la definizione dei requisiti di progettazione per il controllo di tali rischi e la prova che i controlli funzionano come previsto e sono efficaci nell'ambiente designato del dispositivo, garantendo misure di sicurezza sufficienti.
Il "Quadro di sviluppo prodotto sicuro"
Il potenziale danno per il paziente si manifesta quando le minacce alla sicurezza informatica sfruttano le vulnerabilità di un sistema e la facilità con cui queste minacce possono compromettere la sicurezza e l’efficacia di un dispositivo medico aumenta con il numero di vulnerabilità identificate nel tempo. Un Secure Product Development Framework (SPDF) è costituito da processi volti a identificare e ridurre la quantità e la gravità delle vulnerabilità nei prodotti. Comprendendo tutte le fasi del ciclo di vita di un prodotto (progettazione, sviluppo, rilascio, supporto e smantellamento), l'SPDF è parte integrante.
Durante la progettazione del dispositivo, l'integrazione dei processi SPDF può evitare la necessità di una riprogettazione quando si integrano funzionalità basate sulla connettività post-marketing o si affrontano vulnerabilità che comportano rischi incontrollati. La fattibile integrazione con i processi esistenti per lo sviluppo di prodotti e software, la gestione del rischio e il sistema di qualità più ampio aumenta la versatilità dell'SPDF.
Per garantire il rispetto della normativa del Sistema Qualità (SQ) si consiglia l'utilizzo di un SPDF. La FDA incoraggia i produttori ad adottare l’SPDF per i suoi vantaggi nel soddisfare sia il regolamento QS che i requisiti di sicurezza informatica. Si riconosce tuttavia che anche approcci alternativi possono soddisfare il regolamento QS.
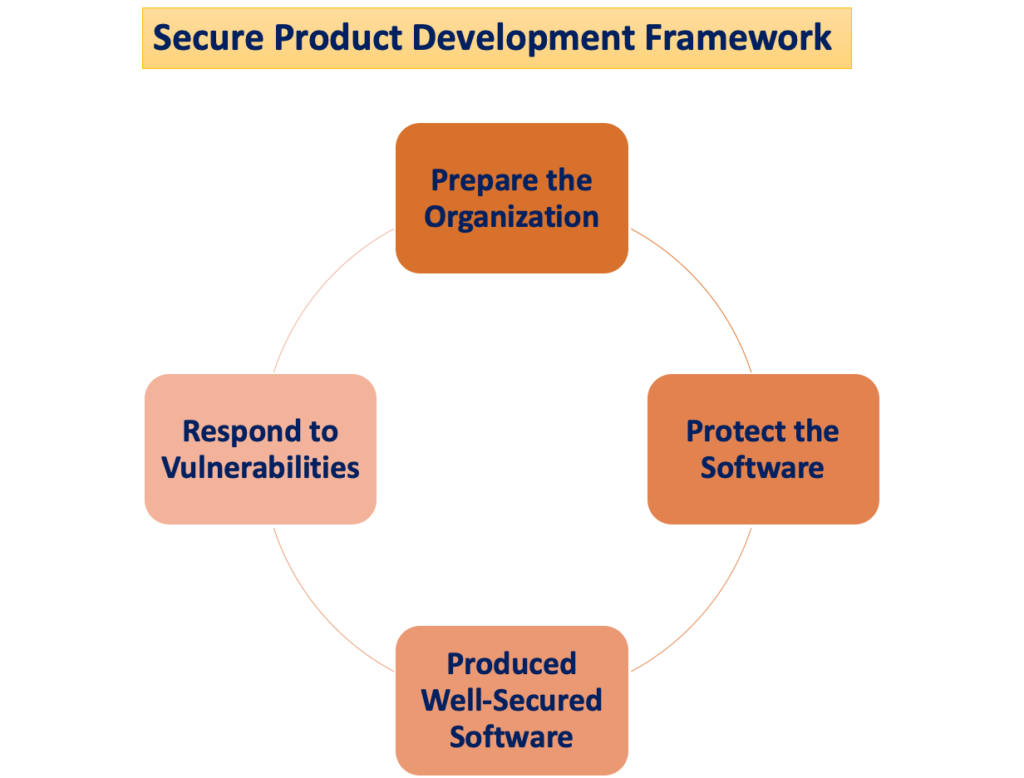
Gestione dei rischi legati alla sicurezza informatica
L'obiettivo principale dell'utilizzo di un SPDF (Secure Product Development Framework) è creare e sostenere dispositivi che siano sicuri ed efficaci. Dal punto di vista della sicurezza, questi dispositivi guadagnano anche affidabilità e resilienza. I produttori e/o gli utenti (ad esempio pazienti, strutture sanitarie) possono quindi gestire questi dispositivi, inclusa l'installazione, la configurazione, gli aggiornamenti e la revisione dei registri dei dispositivi, attraverso la progettazione del dispositivo e l'etichettatura associata.
Le strutture sanitarie hanno la possibilità di gestire questi dispositivi all’interno dei propri quadri di gestione del rischio di sicurezza informatica, come il ampiamente riconosciuto National Institute of Standards and Technology (NIST) Quadro per il miglioramento della sicurezza informatica delle infrastrutture critiche, comunemente denominato Quadro di sicurezza informatica del NIST o NIST CSF.
La FDA raccomanda ai produttori di incorporare processi di progettazione dei dispositivi, come quelli delineati nel regolamento sul sistema di qualità (QS), per rafforzare lo sviluppo e la manutenzione sicuri dei prodotti. Pur mantenendo la flessibilità per i produttori, questi possono anche esplorare quadri alternativi che si allineino al regolamento QS e aderiscano alle raccomandazioni della FDA per l’implementazione di un SPDF. Gli esempi includono il quadro specifico per i dispositivi medici nel Piano di sicurezza congiunto (JSP) per i dispositivi medici e l'informatica sanitaria 30 e IEC 81001-5-1. Anche le normative di altri settori, come ANSI/ISA 62443-4-1 Sicurezza per i sistemi di automazione e controllo industriale Parte 4-1: Requisiti del ciclo di vita dello sviluppo della sicurezza del prodotto, possono soddisfare le normative QS.
Nelle sezioni seguenti di questo articolo vengono fornite raccomandazioni per l'utilizzo dei processi SPDF, come generalmente percepito dalla FDA, che evidenziano considerazioni cruciali per lo sviluppo di dispositivi sicuri ed efficaci. Questi processi integrano il regolamento QS e la FDA suggerisce ai produttori di includere la documentazione corrispondente per la revisione nelle richieste pre-commercializzazione.
Modello di minaccia alla sicurezza informatica
La modellazione delle minacce prevede un processo sistematico per identificare obiettivi di sicurezza, rischi e vulnerabilità in tutto il sistema dei dispositivi medici. Successivamente, comporta la definizione di contromisure per prevenire, mitigare, monitorare o rispondere agli impatti delle minacce durante tutto il ciclo di vita del sistema dei dispositivi medici. Se applicato in modo appropriato e completo, funge da base per ottimizzare la sicurezza di componenti di sistema, prodotti, reti, applicazioni e connessioni.
Per quanto riguarda la gestione dei rischi per la sicurezza e per individuare rischi e controlli adeguati per la sicurezza del sistema dei dispositivi medici, la FDA sostiene l'implementazione della modellazione delle minacce per informare e supportare le attività di analisi dei rischi. Nel contesto della valutazione del rischio, la FDA suggerisce di integrare la modellazione delle minacce durante tutto il processo di progettazione, comprendendo tutti gli elementi del sistema dei dispositivi medici.
Gli aspetti chiave del modello di minaccia dovrebbero comprendere l’identificazione dei rischi e delle misure di mitigazione, informando i rischi pre e post mitigazione considerati nella valutazione del rischio di sicurezza informatica. Inoltre, il modello dovrebbe articolare ipotesi sul sistema dei dispositivi medici o sull’ambiente di utilizzo, ad esempio presupponendo che le reti ospedaliere siano intrinsecamente ostili. Questo presupposto spinge i produttori a considerare scenari in cui un avversario controlla la rete, in grado di alterare, eliminare e riprodurre i pacchetti. Inoltre, il modello di minaccia dovrebbe catturare i rischi di sicurezza informatica introdotti attraverso la catena di fornitura, la produzione, l’implementazione, l’interoperabilità con altri dispositivi, le attività di manutenzione/aggiornamento e le attività di smantellamento, che potrebbero essere trascurati in un tradizionale processo di valutazione dei rischi per la sicurezza.
Per le richieste pre-commercializzazione, la FDA consiglia di includere la documentazione di modellazione delle minacce per mostrare l’analisi del sistema dei dispositivi medici, identificando potenziali rischi per la sicurezza che potrebbero incidere sulla sicurezza e sull’efficacia. I produttori hanno la flessibilità di scegliere tra varie metodologie o combinazioni di metodi per la modellazione delle minacce e la motivazione delle metodologie scelte dovrebbe essere fornita insieme alla documentazione.
Si consiglia di condurre attività di modellazione delle minacce durante le revisioni della progettazione e la documentazione dovrebbe fornire informazioni sufficienti affinché la FDA possa valutare e rivedere le funzionalità di sicurezza integrate nel dispositivo. Questa valutazione olistica dovrebbe considerare sia il dispositivo che il sistema più ampio in cui opera, sottolineando la sicurezza e l’efficacia del dispositivo.
Gli elementi principali dei modelli di minaccia alla cybersecurity possono essere riassunti come segue:
Identificazione di potenziali minacce
Lo sviluppo di un modello di minaccia costituisce un passo fondamentale nel processo di sicurezza informatica, consentendo ai produttori di identificare e comprendere le potenziali minacce alla sicurezza informatica specifiche per i loro dispositivi medici. Lo sviluppo di un modello di minaccia costituisce un passo fondamentale nel processo di sicurezza informatica, consentendo ai produttori di identificare e comprendere le potenziali minacce alla sicurezza informatica specifiche per i loro dispositivi medici. Lo sviluppo di un modello di minaccia costituisce un passo fondamentale nel processo di sicurezza informatica, consentendo ai produttori di identificare e comprendere le potenziali minacce alla sicurezza informatica specifiche per i loro dispositivi medici.
Valutazione e gestione del rischio
La valutazione e la gestione del rischio implicano la valutazione delle minacce identificate per stabilirne il potenziale impatto e l’elaborazione di strategie adeguate per mitigare tali rischi in modo efficace.
Implementazione dei controlli di sicurezza
I controlli di sicurezza sono le misure di salvaguardia o contromisure implementate per proteggere la riservatezza, l'integrità e la disponibilità del dispositivo medico dalle minacce identificate.
Tendenze future nelle linee guida sulla sicurezza informatica della FDA
Con l’evolversi delle minacce e della tecnologia, evolveranno anche le linee guida sulla sicurezza informatica della FDA. È essenziale che i produttori rimangano informati e agili di fronte a questi cambiamenti. I produttori dovrebbero anticipare gli aggiornamenti delle normative rimanendo al passo con le tendenze del settore e mantenendo un approccio proattivo alla sicurezza informatica.
La preparazione per le sfide impreviste è fondamentale; La definizione di solidi protocolli di sicurezza e strategie lungimiranti consentirà ai produttori di rispondere in modo efficace allo stato futuro delle normative sulla sicurezza informatica.
La sicurezza informatica è un aspetto della regolamentazione dei dispositivi medici che non può essere sopravvalutato: è intrinseco alla sicurezza dei dispositivi tanto quanto qualsiasi caratteristica meccanica o elettrica. La FDA lo ha riconosciuto e, implementando un quadro completo di sicurezza informatica, mira a tenere il passo con l’innovazione proteggendo al contempo la salute pubblica. Produttori, operatori sanitari e pazienti devono collaborare per sostenere questi standard e garantire la continua affidabilità e sicurezza dei dispositivi medici di fronte alle minacce informatiche.
Iscriviti alla newsletter di QualityMedDev
QualityMedDev è una piattaforma online focalizzata su argomenti di qualità e regolamentazione per il business dei dispositivi medici; Seguici su LinkedIn ed Twitter per essere sempre aggiornato sulle più importanti novità in ambito normativo.
QualityMedDev è una delle più grandi piattaforme online a supporto del business dei dispositivi medici per argomenti di conformità normativa. Noi forniamo servizi di consulenza normativa su una vasta gamma di argomenti, da UE MDR e IVDR a ISO 13485, inclusa la gestione del rischio, la biocompatibilità, l'usabilità e la verifica e validazione del software e, in generale, il supporto nella preparazione della documentazione tecnica per l'MDR.
La nostra piattaforma sorella Accademia QualityMedDev offre la possibilità di seguire corsi di formazione online e di autoapprendimento focalizzati su temi di compliance normativa per i dispositivi medici. Questi corsi di formazione, sviluppati in collaborazione con professionisti altamente qualificati nel settore dei dispositivi medici, ti consentono di aumentare in modo esponenziale le tue competenze su un'ampia gamma di argomenti di qualità e normativi per le operazioni di business dei dispositivi medici.

Non esitare ad iscriverti alla nostra Newsletter!
- Distribuzione di contenuti basati su SEO e PR. Ricevi amplificazione oggi.
- PlatoData.Network Generativo verticale Ai. Potenzia te stesso. Accedi qui.
- PlatoAiStream. Intelligenza Web3. Conoscenza amplificata. Accedi qui.
- PlatoneESG. Carbonio, Tecnologia pulita, Energia, Ambiente, Solare, Gestione dei rifiuti. Accedi qui.
- Platone Salute. Intelligence sulle biotecnologie e sulle sperimentazioni cliniche. Accedi qui.
- Fonte: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- :ha
- :È
- :non
- :Dove
- $ SU
- 2016
- 30
- 350
- 820
- 9
- a
- WRI
- Academy
- riconosciuto
- operanti in
- attività
- Inoltre
- indirizzo
- indirizzamento
- Aggiunge
- aderire
- amministrazione
- consigliato
- sostenitori
- agile
- Mirato
- mira
- allineare
- Tutti
- consente
- anche
- alternativa
- an
- analisi
- .
- ed
- anticipare
- in qualsiasi
- applicabile
- applicazioni
- applicato
- APPLICA
- approccio
- approcci
- opportuno
- appropriatamente
- SONO
- articolo
- AS
- aspetto
- aspetti
- valutare
- valutazione
- associato
- assunzione
- ipotesi
- garanzia
- At
- attacchi
- Automatizzata
- Automazione
- disponibilità
- BE
- diventare
- diventando
- stato
- vantaggi
- sostenere
- entrambi
- ampio
- più ampia
- portato
- affari
- operazioni affaristiche
- ma
- by
- Materiale
- non può
- capace
- catturare
- certo
- catena
- sfide
- Modifiche
- Scegli
- scelto
- classi
- collaboreranno
- collaborazione
- COM
- combinazioni
- comunemente
- Complemento
- conformità
- componenti
- globale
- compromesso
- concezione
- preoccupazioni
- riservatezza
- Configurazione
- collegato
- Dispositivi collegati
- Connessioni
- Prendere in considerazione
- Considerazioni
- considerato
- coerente
- consiste
- costituire
- consulting
- musica
- contesto
- continua
- di controllo
- controllo
- controlli
- Corrispondente
- potuto
- paesi
- Corsi
- copertura
- creare
- critico
- Infrastruttura critica
- cruciale
- Cyber
- Attacchi informatici
- Cybersecurity
- Data
- definizione
- definizione
- dimostrando
- deployment
- Design
- processo di progettazione
- designato
- determinazione
- sviluppato
- in via di sviluppo
- Mercato
- dispositivo
- dispositivi
- diverso
- digitale
- diminuzione
- discutere
- documentazione
- lancio
- droga
- durante
- e
- più presto
- guadagnare
- alleviare
- Efficace
- in maniera efficace
- efficacia
- o
- elementi
- abbraccio
- enfatizzando
- impiegando
- consentendo
- circondare
- comprende
- che comprende
- incoraggia
- garantire
- assicurando
- Ambiente
- essential
- stabilire
- sviluppate
- stabilire
- Etere (ETH)
- EU
- europeo
- Unione europea
- la valutazione
- valutazione
- EVER
- prova
- evolvere
- Esempi
- esistente
- Sfruttare
- esplora
- in modo esponenziale
- Faccia
- strutture
- fda
- fattibile
- caratteristica
- Caratteristiche
- campo
- Flessibilità
- concentrato
- seguire
- i seguenti
- segue
- cibo
- Food and Drug Administration
- Food and Drug Administration (FDA)
- Nel
- prima linea
- lungimirante
- essere trovato
- Fondazione
- Fondamentale
- Contesto
- quadri
- da
- pieno
- funzionalità
- Inoltre
- futuro
- Generale
- generalmente
- linee guida
- nuocere
- Avere
- Salute e benessere
- assistenza sanitaria
- Alta
- Highlight
- vivamente
- olistica
- Ospedale
- Tuttavia
- HTML
- HTTPS
- Identificazione
- identificato
- identificare
- identificazione
- if
- Impact
- impatti
- implementazione
- implementato
- Implementazione
- importante
- miglioramento
- in
- includere
- inclusi
- Compreso
- incorporare
- incorporando
- Aumento
- Aumenta
- sempre più
- industriale
- automazione industriale
- industria
- far sapere
- informazioni
- informati
- Infrastruttura
- intrinsecamente
- inizialmente
- Innovazione
- installazione
- esempio
- Istituto
- integrale
- integrato
- Integrazione
- integrazione
- interezza
- destinato
- ai miglioramenti
- intrinseco
- introdotto
- coinvolgere
- comporta
- ISO
- IT
- SUO
- giunto
- mantenere
- Le
- etichettatura
- maggiore
- ciclo vitale
- ciclo di vita
- MailChimp
- Principale
- mantenere
- mantenimento
- manutenzione
- gestire
- gestione
- mandati
- Produttori
- consigliato per la
- max-width
- Maggio..
- MDR
- analisi
- meccanico
- medicale
- dispositivo medico
- dispositivi medici
- Soddisfare
- incontro
- metodologie
- metodi
- forza
- Ridurre la perdita dienergia con una
- attenuante
- mitigazione dei rischi
- modello
- modellismo
- modelli
- Monitorare
- Scopri di più
- maggior parte
- devono obbligatoriamente:
- il
- Natura
- Navigazione
- necessità
- Bisogno
- Rete
- reti
- notizie
- nista
- numero
- obiettivo
- Obiettivi d'Esame
- of
- on
- ONE
- online
- esclusivamente
- operare
- opera
- Operazioni
- ottimizzazione
- Opzione
- or
- Altro
- nostro
- delineato
- uscite
- ancora
- svista
- proprio
- Pace
- pacchetti
- parte
- paziente
- pazienti
- percepito
- prospettiva
- fase
- centrale
- piano
- piattaforma
- Platone
- Platone Data Intelligence
- PlatoneDati
- gioca
- plug-in
- posto
- posizione
- possibilità
- Post
- potenziale
- pratiche
- preparazione
- prevenire
- primario
- Proactive
- procedure
- processi
- i processi
- Prodotto
- sviluppo del prodotto
- Prodotti
- Scelto dai professionisti
- istruzioni
- protegge
- proteggere
- protocolli
- fornire
- purché
- fornisce
- fornitura
- la percezione
- sanità pubblica
- pubblicato
- qualità
- quantità
- gamma
- fondamento logico
- ragionevole
- riconosciuto
- raccomandazioni
- raccomandato
- raccomanda
- di cui
- Regolamento
- normativa
- normativo
- Conformità normativa
- relazionato
- relazione
- rilasciare
- problemi di
- rimanere
- necessario
- Requisiti
- elasticità
- Rispondere
- recensioni
- Recensioni
- Aumento
- Rischio
- valutazione del rischio
- gestione del rischio
- rischi
- robusto
- Ruolo
- sicura
- salvaguardare
- garanzie
- Sicurezza
- stesso
- Scenari
- sezioni
- settore
- Settori
- sicuro
- problemi di
- Misure di sicurezza
- gestione dei rischi per la sicurezza
- rischi per la sicurezza
- serve
- alcuni
- gravità
- dovrebbero
- vetrina
- sorella
- qualificato
- più intelligente
- So
- Software
- lo sviluppo del software
- venduto
- specifico
- Specifiche tecniche
- specificato
- Stage
- tappe
- standard
- Regione / Stato
- stati
- soggiorno
- soggiorno
- step
- strategie
- sottomissione
- Articoli
- sottoscrivi
- Successivamente
- tale
- sufficiente
- suggerisce
- adatto
- fornire
- supply chain
- supporto
- Supporto
- sistema
- SISTEMI DI TRATTAMENTO
- Consulenza
- Tecnologia
- di
- che
- Il
- Il futuro
- loro
- poi
- Strumenti Bowman per analizzare le seguenti finiture:
- di
- questo
- quelli
- minaccia
- minacce
- Attraverso
- per tutto
- tempo
- volte
- a
- pure
- Argomenti
- tradizionale
- Training
- tendenze
- Affidati ad
- attendibilità
- capire
- imprevisto
- unione
- Unito
- Stati Uniti
- Aggiornamenti
- sostenere
- URL
- us
- usabilità
- Impiego
- uso
- utenti
- Utilizzando
- convalidando
- convalida
- vario
- Convalida
- versatilità
- vulnerabilità
- Vulnerabile
- we
- siti web
- quando
- se
- quale
- while
- ampiamente
- volere
- con
- entro
- WordPress
- WordPress Plugin
- Tu
- Trasferimento da aeroporto a Sharm
- zefiro




