Toidu- ja ravimiamet (FDA) mängib keskset rolli rahvatervise kaitsmisel meditsiiniseadmete reguleerimise kaudu, tagades, et need on ettenähtud kasutuse korral ohutud ja tõhusad. Tänapäeval on ühendatud seadmete kasv toonud küberjulgeoleku FDA murede esiplaanile; selles kontekstis on küberjulgeolekuohu mudeli määratlemiseks konkreetsete nõuete määratlemine hädavajalik. Kuna meditsiiniseadmed integreeritakse üha enam tehnoloogiaga, on vajadus tugevate küberjulgeolekumeetmete järele patsiendi teabe kaitsmiseks ja nende seadmete funktsionaalsuse tagamiseks kriitilisem kui kunagi varem. FDA eeskirjade järgimine ei ole oluline mitte ainult tootjate vastavuse tagamiseks, vaid ka patsientide ja kasutajate ohutuse ja usalduse tagamiseks.
Oleme QualityMedDevi veebisaitidel mitu korda arutanud digitaalsete meditsiiniseadmete ja nendega seotud nõuete üle, käsitledes erinevaid teemasid, näiteks AI meditsiiniseadmetes, digitaalsed tervisetootedja TGA lähenemine digitaalsete meditsiiniseadmete reguleerimiseks. Samal ajal on FDA avaldanud erinevad juhised seoses küberturvalisusega, kas seoses turustamiseelsed nõuded ja turustamisjärgsed nõuded.
FDA on kehtestanud küberturvalisuse nõuded osana oma regulatiivsest järelevalvest, et kaitsta patsiente ja tagada meditsiiniseadmete terviklikkus. Need standardid kehtivad seadme kogu elutsükli kohta alates esialgsest projekteerimisest ja arendusest kuni juurutamise ja hoolduseni. Meditsiiniseadmed muutuvad üha nutikamaks ja ühendatumaks, mistõttu on need küberohtude suhtes üha haavatavamad. Küberturvalisuse tavade integreerimine meditsiiniseadmete arendusfaasis on küberrünnakutest tulenevate riskide maandamisel otsustava tähtsusega samm.
FDA küberturvalisuse raamistiku põhikomponendid
Meditsiiniseadmete küberjulgeoleku terviklikkuse tagamiseks on FDA välja toonud turustamiseelsed nõuded, mis hõlmavad tootjate vajadust lisada küberjulgeolekuohu mudel ja turbekontroll seadme väljatöötamise kõige varasemast etapist alates.
Seadmete tootjad kehtestavad kvaliteedisüsteemid ja järgivad neid, et tagada nende toodete järjepidev vastavus kohaldatavatele nõuetele ja spetsifikatsioonidele. Nende kvaliteedisüsteemide nõuded leiate kvaliteedisüsteemi (QS) määrusest 21 CFR Part 820, kui seadet müüakse Ameerika Ühendriikides, või muudest eeskirjadest teiste riikide jaoks (nt EU MDR 2017/745 Euroopa Liidu jaoks).
Olenevalt seadme olemusest võivad QS-i nõuded olla asjakohased müügieelsel, turustamisjärgsel etapil või mõlemal. Turustamiseelse etapi kontekstis võib teatud küberjulgeolekuriskidega seadmete ohutuse ja tõhususe mõistliku kindluse näitamine hõlmata QS-i määrusega seotud dokumentatsiooni väljundite lisamist turustamiseelsesse esitamisse. Näiteks nõuavad ISO 13485:2016 ja 21 CFR 820, et kõikide tarkvaraga automatiseeritud seadmete klasside tootjad kehtestavad protseduurid seadme konstruktsiooni kontrollimiseks, tagades vastavuse kindlaksmääratud konstruktsiooninõuetele (edaspidi "disaini juhtelemendid"). Disainikontrolli raames peavad tootjad kehtestama ja säilitama protseduurid seadme disaini kinnitamiseks, mis hõlmab vajaduse korral tarkvara valideerimist ja riskianalüüsi. Tarkvara kohustusliku valideerimise ja riskianalüüsi osana võivad tarkvaraseadmete tootjad vajaduse korral käivitada küberturvalisuse riskijuhtimis- ja valideerimisprotsessid.
Tarkvara valideerimine ja riskijuhtimine on küberjulgeoleku analüüside olulised elemendid, määrates kindlaks, kas seade tagab piisava ohutuse ja tõhususe. FDA annab tootjatele ülesandeks lisada projekteerimiskontrolli osana arendusprotsesse, mis arvestavad ja käsitlevad tarkvarariske kogu projekteerimise ja arendusetapis. Need protsessid peaksid hõlmama küberturvalisuse kaalutlusi. See hõlmab turvariskide tuvastamisega tegelemist, nende riskide ohjamiseks disaininõuete kehtestamist ja tõendite esitamist selle kohta, et juhtelemendid toimivad ettenähtud viisil ja on tõhusad seadme määratud keskkonnas, tagades piisavad turvameetmed.
"Turvaline tootearenduse raamistik"
Patsientide kahjustamise potentsiaal tekib siis, kui küberjulgeolekuohud kasutavad ära süsteemi haavatavusi, ning see, kui lihtne need ohud võivad meditsiiniseadme ohutust ja tõhusust kahjustada, suureneb aja jooksul tuvastatud haavatavuste arvuga. Turvaline tootearenduse raamistik (SPDF) koosneb protsessidest, mille eesmärk on tuvastada ja vähendada toodete haavatavuste hulka ja raskusastet. SPDF, mis hõlmab toote elutsükli kõiki etappe – projekteerimine, arendus, väljalaskmine, tugi ja kasutusest kõrvaldamine – on lahutamatu osa.
Seadme kavandamise ajal võib SPDF-protsesside kaasamine takistada turustamisjärgsete ühenduvuspõhiste funktsioonide integreerimisel või kontrollimatuid riske kujutavate haavatavuste kõrvaldamisel ümberkujundamise vajadust. Teostatav integreerimine olemasolevate protsessidega toote- ja tarkvaraarenduse, riskijuhtimise ja laiema kvaliteedisüsteemiga suurendab SPDF-i mitmekülgsust.
Kvaliteedisüsteemi (QS) määruse järgimise tagamiseks on soovitatav kasutada SPDF-i. FDA julgustab tootjaid SPDF-i omaks võtma, kuna see toob kasu nii QS-määruse kui ka küberturvalisuse nõuete täitmisel. Siiski tunnistatakse, et ka alternatiivsed lähenemisviisid võivad QS määrusele vastata.
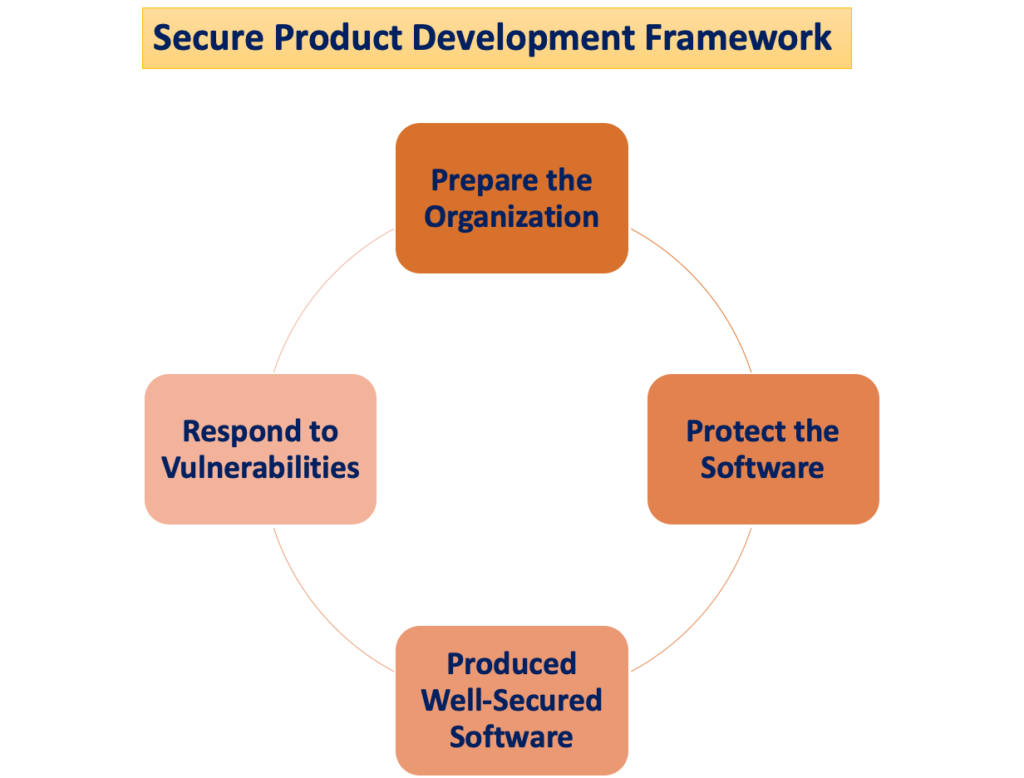
Küberjulgeolekuriskide juhtimine
SPDF-i (Secure Product Development Framework) kasutamise peamine eesmärk on luua ja säilitada seadmeid, mis on nii ohutud kui ka tõhusad. Turvalisuse seisukohast teenivad need seadmed ka usaldusväärsust ja vastupidavust. Tootjad ja/või kasutajad (nt patsiendid, tervishoiuasutused) saavad seejärel neid seadmeid hallata, sealhulgas installida, konfigureerida, värskendada ja seadmete logisid üle vaadata, kasutades seadme disaini ja sellega seotud märgistusi.
Tervishoiuasutustel on võimalus hallata neid seadmeid oma küberturvalisuse riskijuhtimise raamistikes, näiteks laialdaselt tunnustatud riiklikus standardite ja tehnoloogia instituudis (NIST) Kriitilise infrastruktuuri küberturvalisuse parandamise raamistik, mida tavaliselt nimetatakse NIST küberturvalisuse raamistik või NIST CSF.
FDA soovitab tootjatel kaasata seadmete projekteerimisprotsesse, nagu need, mis on kirjeldatud kvaliteedisüsteemi (QS) määruses, et toetada turvalist tootearendust ja hooldust. Säilitades tootjate jaoks paindlikkuse, võivad nad uurida ka alternatiivseid raamistikke, mis on vastavuses QS-määrusega ja järgivad FDA soovitusi SPDF-i rakendamiseks. Näited hõlmavad meditsiiniseadmete ja tervishoiu IT ühises turbeplaanis (JSP) 30 sisalduvat meditsiiniseadme spetsiifilist raamistikku ja IEC 81001-5-1. Teiste sektorite raamistikud, nagu ANSI/ISA 62443-4-1 tööstusautomaatika ja juhtimissüsteemide turvalisus Osa 4-1: Toote turvalisuse arendamise elutsükli nõuded, võivad samuti vastata QS eeskirjadele.
Selle artikli järgmistes osades on esitatud soovitused SPDF-protsesside kasutamiseks, nagu FDA üldiselt tajub, mis tõstavad esile olulised kaalutlused ohutute ja tõhusate seadmete väljatöötamisel. Need protsessid täiendavad QS-i määrust ja FDA soovitab tootjatel lisada turustamiseelsesse esildistesse ülevaatamiseks vastavad dokumendid.
Küberjulgeolekuohu mudel
Ohtude modelleerimine hõlmab süstemaatilist protsessi turvaeesmärkide, riskide ja haavatavuste tuvastamiseks kogu meditsiiniseadmete süsteemis. Seejärel hõlmab see vastumeetmete määratlemist ohtude mõju ennetamiseks, leevendamiseks, jälgimiseks või neile reageerimiseks kogu meditsiiniseadme süsteemi elutsükli jooksul. Asjakohase ja tervikliku rakendamise korral on see aluseks süsteemi komponentide, toodete, võrkude, rakenduste ja ühenduste turvalisuse optimeerimisele.
Seoses turvariskide juhtimisega ning meditsiiniseadmete süsteemi jaoks sobivate turvariskide ja kontrollide kindlaksmääramisega toetab FDA ohumudelite rakendamist, et teavitada ja toetada riskianalüüsi tegevusi. Riskianalüüsi kontekstis soovitab FDA integreerida ohtude modelleerimise kogu projekteerimisprotsessi, hõlmates kõiki meditsiiniseadmete süsteemi elemente.
Ohumudeli põhiaspektid peaksid hõlmama riskide tuvastamist ja leevendamist, teavitades nii enne kui ka pärast küberjulgeoleku riskide hindamisel arvesse võetud riskidest. Lisaks peaks mudel sõnastama eeldused meditsiiniseadmete süsteemi või kasutuskeskkonna kohta, näiteks eeldades, et haiglavõrgud on oma olemuselt vaenulikud. See eeldus sunnib tootjaid kaaluma stsenaariume, kus vastane kontrollib võrku, olles võimeline pakette muutma, maha viskama ja uuesti esitama. Lisaks peaks ohumudel hõlmama küberjulgeoleku riske, mis tekivad tarneahelas, tootmises, juurutamises, koostoimimises teiste seadmetega, hooldus-/uuendustegevustes ja dekomisjoneerimistoimingutes, mida võib traditsioonilises ohutusriskide hindamise protsessis tähelepanuta jätta.
Turueelsete esildiste puhul soovitab FDA lisada ohtude modelleerimise dokumentatsiooni, et tutvustada meditsiiniseadmete süsteemi analüüsi, tuvastades võimalikud turvariskid, mis võivad mõjutada ohutust ja tõhusust. Tootjatel on ohu modelleerimiseks võimalik paindlikult valida erinevate metoodikate või meetodite kombinatsioonide vahel ning nende valitud metoodikate põhjendus tuleks esitada koos dokumentatsiooniga.
Disainiülevaatuste ajal on soovitatav läbi viia ohtude modelleerimise tegevusi ning dokumentatsioon peaks andma FDA-le piisavalt teavet seadmesse integreeritud turvaelementide hindamiseks ja ülevaatamiseks. See terviklik hindamine peaks võtma arvesse nii seadet kui ka laiemat süsteemi, milles see töötab, rõhutades seadme ohutust ja tõhusust.
Küberjulgeolekuohu mudelite põhielemendid võib kokku võtta järgmiselt:
Võimalike ohtude tuvastamine
Ohumudeli väljatöötamine on küberturvalisuse protsessi põhietapp, mis võimaldab tootjatel tuvastada ja mõista nende meditsiiniseadmetele omaseid võimalikke küberjulgeoleku ohte. Ohumudeli väljatöötamine on küberturvalisuse protsessi põhietapp, mis võimaldab tootjatel tuvastada ja mõista nende meditsiiniseadmetele omaseid võimalikke küberjulgeoleku ohte. Ohumudeli väljatöötamine on küberturvalisuse protsessi põhietapp, mis võimaldab tootjatel tuvastada ja mõista nende meditsiiniseadmetele omaseid võimalikke küberjulgeoleku ohte.
Riski hindamine ja juhtimine
Riskide hindamine ja juhtimine hõlmavad tuvastatud ohtude hindamist, et teha kindlaks nende potentsiaalne mõju, ja asjakohaste strateegiate väljatöötamist nende riskide tõhusaks maandamiseks.
Turvakontrolli rakendamine
Turvakontrollid on kaitse- või vastumeetmed, mida rakendatakse meditsiiniseadme konfidentsiaalsuse, terviklikkuse ja kättesaadavuse kaitsmiseks tuvastatud ohtude eest.
Tulevased suundumused FDA küberturvalisuse juhistes
Ohtude ja tehnoloogia arenedes muutuvad ka FDA küberjulgeoleku juhised. On oluline, et tootjad oleksid nende muudatustega silmitsi seistes kursis ja paindlikud. Tootjad peaksid ennetama eeskirjade värskendusi, hoides end kursis valdkonna suundumustega ja säilitades küberturvalisusele ennetava lähenemisviisi.
Ettenägematuteks väljakutseteks valmistumine on võtmetähtsusega; tugevate turvaprotokollide ja tulevikku suunatud strateegiate loomine võimaldab tootjatel tõhusalt reageerida küberjulgeoleku eeskirjade tulevastele oludele.
Küberturvalisus on meditsiiniseadmete reguleerimise aspekt, mida ei saa üle tähtsustada – see on seadme ohutusele sama oluline kui mis tahes mehaaniline või elektriline funktsioon. FDA on seda tunnistanud ja rakendades kõikehõlmavat küberjulgeoleku raamistikku, püüab ta innovatsiooniga sammu pidada, kaitstes samal ajal rahvatervist. Tootjad, tervishoiutöötajad ja patsiendid peavad tegema koostööd, et järgida neid standardeid ning tagada meditsiiniseadmete jätkuv töökindlus ja ohutus küberohtude korral.
Tellige QualityMedDevi uudiskiri
QualityMedDev on veebiplatvorm, mis keskendub meditsiiniseadmete äri kvaliteedi- ja regulatiivteemadele; Jälgi meid LinkedIn ja puperdama et olla kursis kõige olulisemate uudistega reguleerimise valdkonnas.
QualityMedDev on üks suurimaid veebiplatvorme, mis toetab meditsiiniseadmete äritegevust eeskirjade järgimise teemadel. Me varustame regulatiivsed konsultatsiooniteenused paljudel teemadel, alates EU MDR & IVDR et ISO 13485, sealhulgas riskijuhtimine, biosobivus, kasutatavus ja tarkvara kontrollimine ja valideerimine ning üldiselt tugi MDR-i tehnilise dokumentatsiooni koostamisel.
Meie sõsarplatvorm QualityMedDevi akadeemia annab võimaluse jälgida veebipõhiseid ja iseseisva tempoga koolitusi, mis keskenduvad meditsiiniseadmete eeskirjadele vastavuse teemadele. Need koolitused, mis on välja töötatud koostöös kõrgelt kvalifitseeritud meditsiiniseadmete sektori spetsialistidega, võimaldavad teil plahvatuslikult suurendada oma pädevusi paljudel meditsiiniseadmete äritegevuse kvaliteedi- ja regulatiivsetel teemadel.

Ärge kõhelge meie uudiskirja tellimisest!
- SEO-põhise sisu ja PR-levi. Võimenduge juba täna.
- PlatoData.Network Vertikaalne generatiivne Ai. Jõustage ennast. Juurdepääs siia.
- PlatoAiStream. Web3 luure. Täiustatud teadmised. Juurdepääs siia.
- PlatoESG. Süsinik, CleanTech, Energia, Keskkond päikeseenergia, Jäätmekäitluse. Juurdepääs siia.
- PlatoTervis. Biotehnoloogia ja kliiniliste uuringute luureandmed. Juurdepääs siia.
- Allikas: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- :on
- :on
- :mitte
- : kus
- $ UP
- 2016
- 30
- 350
- 820
- 9
- a
- MEIST
- Akadeemia
- tunnustatud
- üle
- tegevus
- Lisaks
- aadress
- adresseerimine
- Lisab
- kinni pidama
- haldamine
- soovitatav
- toetajad
- väle
- eesmärk
- Eesmärgid
- viia
- Materjal: BPA ja flataatide vaba plastik
- võimaldab
- Ka
- alternatiiv
- an
- analüüsid
- analüüs
- ja
- ennetada
- mistahes
- kohaldatav
- rakendused
- rakendatud
- kehtima
- lähenemine
- lähenemisviisid
- asjakohane
- asjakohaselt
- OLEME
- artikkel
- AS
- aspekt
- aspektid
- hinnata
- hindamine
- seotud
- eeldus
- eeldused
- kinnitus
- At
- Reageerib
- Automatiseeritud
- Automaatika
- kättesaadavus
- BE
- muutuma
- saada
- olnud
- Kasu
- toetama
- mõlemad
- lai
- laiem
- tõi kaasa
- äri
- äritegevus
- kuid
- by
- CAN
- ei saa
- võimeline
- lüüa
- kindel
- kett
- väljakutseid
- Vaidluste lahendamine
- Vali
- valitud
- klassid
- Teevad koostööd
- koostöö
- COM
- kombinatsioonid
- tavaliselt
- Täiendus
- Vastavus
- komponendid
- terviklik
- kompromiss
- disain
- Murettekitav
- konfidentsiaalsus
- konfiguratsioon
- seotud
- ühendatud seadmeid
- Side
- Arvestama
- kaalutlused
- kaaluda
- järjepidev
- koosneb
- moodustama
- nõustamine
- kaasaegne
- kontekst
- jätkas
- kontrollida
- kontroll
- kontrolli
- Vastav
- võiks
- riikides
- kursused
- kattes
- looma
- kriitiline
- Kriitiline infrastruktuur
- otsustav
- cyber
- Küberrünnakud
- Küberturvalisus
- kuupäev
- määratlemisel
- määratlus
- näidates
- kasutuselevõtu
- Disain
- projekteerimisprotsess
- määratud
- määrates kindlaks
- arenenud
- arenev
- & Tarkvaraarendus
- seade
- seadmed
- erinev
- digitaalne
- väheneb
- arutame
- dokumentatsioon
- Kukkumine
- uimasti
- ajal
- e
- varem
- teenima
- leevendada
- Tõhus
- tõhusalt
- tõhusus
- kumbki
- elemendid
- omaks võtma
- rõhutades
- tööle
- võimaldades
- hõlmab
- hõlmab
- haarav
- julgustab
- tagama
- tagades
- keskkond
- oluline
- looma
- asutatud
- asutades
- Eeter (ETH)
- EU
- Euroopa
- Euroopa Liit
- hindamine
- hindamine
- KUNAGI
- tõend
- arenema
- näited
- olemasolevate
- Ekspluateeri
- uurima
- eksponentsiaalselt
- nägu
- rajatised
- FDA
- teostatav
- tunnusjoon
- FUNKTSIOONID
- väli
- Paindlikkus
- keskendunud
- järgima
- Järel
- järgneb
- toit
- Food and Drug Administration
- Toidu- ja ravimiamet (FDA)
- eest
- esirinnas
- tulevikku
- avastatud
- Sihtasutus
- Sihtasutus
- Raamistik
- raamistikud
- Alates
- täis
- funktsionaalsus
- Pealegi
- tulevik
- Üldine
- üldiselt
- suunised
- kahju
- Olema
- Tervis
- tervishoid
- Suur
- Esile tõstma
- kõrgelt
- terviklikku
- Haigla
- aga
- HTML
- HTTPS
- Identifitseerimine
- tuvastatud
- identifitseerima
- identifitseerimiseks
- if
- mõju
- Mõjud
- täitmine
- rakendatud
- rakendamisel
- oluline
- Paranemist
- in
- sisaldama
- hõlmab
- Kaasa arvatud
- lisada
- kaasates
- Suurendama
- Tõstab
- üha rohkem
- tööstus-
- tööstusautomaatika
- tööstus
- teatama
- info
- teavitatakse
- Infrastruktuur
- olemuselt
- esialgne
- Innovatsioon
- paigaldamine
- Näiteks
- Instituut
- lahutamatu
- integreeritud
- Integreerimine
- integratsioon
- terviklikkuse
- ette nähtud
- sisse
- sisemine
- sisse
- kaasama
- hõlmab
- ISO
- IT
- ITS
- ühine
- hoidma
- Võti
- märgistamine
- suurim
- eluring
- eluring
- MailChimpi
- põhiline
- säilitada
- säilitamine
- hooldus
- juhtima
- juhtimine
- mandaadid
- Tootjad
- tootmine
- max laiuse
- mai..
- MDR
- meetmed
- mehaaniline
- meditsiini-
- Meditsiiniline seade
- meditsiiniseadmete
- Vastama
- koosolekul
- metoodikad
- meetodid
- võib
- Leevendada
- leevendav
- riskide maandamine
- mudel
- modelleerimine
- mudelid
- Jälgida
- rohkem
- kõige
- peab
- riiklik
- loodus
- NAVIGATSIOON
- vajadus
- Vajadus
- võrk
- võrgustikud
- uudised
- nst
- number
- eesmärk
- eesmärgid
- of
- on
- ONE
- Internetis
- ainult
- töötama
- tegutseb
- Operations
- optimeerimine
- valik
- or
- Muu
- meie
- välja toodud
- väljundid
- üle
- Järelevalve
- enda
- tempo
- paketid
- osa
- patsient
- patsientidel
- tajutav
- perspektiiv
- faas
- Keskses
- kava
- inimesele
- Platon
- Platoni andmete intelligentsus
- PlatoData
- mängib
- plugin
- tulenevad
- positsioon
- võimalus
- Postitusi
- potentsiaal
- tavad
- ettevalmistamine
- vältida
- esmane
- Proaktiivne
- menetlused
- protsess
- Protsessid
- Toode
- tootearendus
- Toodet
- spetsialistid
- küsib
- kaitsma
- kaitsta
- protokollid
- anda
- tingimusel
- annab
- pakkudes
- avalik
- rahvatervise
- avaldatud
- kvaliteet
- kogus
- valik
- põhjendus
- mõistlik
- tunnustatud
- soovitused
- soovitatav
- soovitab
- nimetatud
- Määrus
- määrused
- regulatiivne
- nõuetele vastavuse
- seotud
- seos
- vabastama
- usaldusväärsus
- jääma
- nõutav
- Nõuded
- vastupidavust
- Reageerida
- läbi
- Arvustused
- Tõusma
- Oht
- riskianalüüsi
- riskijuhtimise
- riskide
- jõuline
- Roll
- ohutu
- kaitsmine
- tagatisi
- ohutus
- sama
- stsenaariumid
- lõigud
- sektor
- Sektorid
- kindlustama
- turvalisus
- Turvameetmed
- turvariskide juhtimine
- turvariskid
- teenib
- mitu
- ägedus
- peaks
- presentatsioon
- õde
- osav
- targemaks
- So
- tarkvara
- tarkvaraarenduse
- müüdud
- konkreetse
- spetsifikatsioonid
- määratletud
- Stage
- etappidel
- standardite
- riik
- Ühendriigid
- jääma
- viibides
- Samm
- strateegiad
- esitamine
- Ettepanekud
- tellima
- Järgnevalt
- selline
- piisav
- Soovitab
- sobiv
- varustama
- tarneahelas
- toetama
- Toetamine
- süsteem
- süsteemid
- Tehniline
- Tehnoloogia
- kui
- et
- .
- Tulevik
- oma
- SIIS
- Need
- nad
- see
- need
- oht
- ähvardused
- Läbi
- läbi kogu
- aeg
- korda
- et
- liiga
- Teemasid
- traditsiooniline
- koolitus
- Trends
- Usalda
- usaldusväärsus
- mõistma
- ettenägematu
- liit
- Ühendatud
- Ühendriigid
- Uudised
- Üles
- URL
- us
- kasutatavus
- Kasutus
- kasutama
- Kasutajad
- kasutades
- valideerimine
- kinnitamine
- eri
- Kontrollimine
- mitmekülgsus
- Haavatavused
- Haavatav
- we
- veebilehed
- millal
- kas
- mis
- kuigi
- laialdaselt
- will
- koos
- jooksul
- WordPress
- WordPress Plugin
- sa
- Sinu
- sephyrnet





