Food and Drug Administration (FDA) spiller en central rolle i at beskytte folkesundheden gennem regulering af medicinsk udstyr, der sikrer, at de er både sikre og effektive til deres tilsigtede brug. I nutidige tider har fremkomsten af tilsluttede enheder bragt cybersikkerhed på forkant med FDA-bekymringer; i denne sammenhæng er det afgørende at definere specifikke krav til definitionen af en trusselsmodel for cybersikkerhed. Efterhånden som medicinsk udstyr bliver mere og mere integreret med teknologi, er behovet for robuste cybersikkerhedsforanstaltninger til at beskytte patientoplysninger og sikre funktionaliteten af disse enheder mere kritisk end nogensinde. Overholdelse af FDA-regler er ikke kun afgørende for fabrikanternes overensstemmelse, men også for patienternes og brugernes sikkerhed og tillid.
Vi har diskuteret digitalt medicinsk udstyr og relaterede krav flere gange på QualityMedDev-websteder, der dækker forskellige emner som f.eks. AI inden for medicinsk udstyr, digitale sundhedsprodukterog TGA tilgang til regulering af digitalt medicinsk udstyr. Samtidig har FDA udgivet forskellige retningslinjer i forhold til cybersikkerhed, enten ift krav før markedet , krav efter markedsføring.
FDA har etableret cybersikkerhedskrav som en del af deres lovgivningsmæssige tilsyn for at beskytte patienter og sikre integriteten af medicinsk udstyr. Disse standarder gælder for en enheds fulde livscyklus, fra indledende design og udvikling til implementering og vedligeholdelse. Med medicinsk udstyr, der bliver smartere og mere forbundet, er de i stigende grad sårbare over for cybertrusler. Integrationen af cybersikkerhedspraksis i udviklingsfasen af medicinsk udstyr er et afgørende skridt for at afbøde risici forbundet med cyberangreb.
Nøglekomponenter i FDA's Cybersecurity Framework
For at sikre cybersikkerhedsintegriteten af medicinsk udstyr har FDA skitseret krav før markedet, som omfatter behovet for, at producenter skal inkorporere cybersikkerhedstrusselmodeller og sikkerhedskontroller fra de tidligste stadier af enhedens udformning.
Producenter af udstyr skal etablere og overholde kvalitetssystemer for at sikre ensartet overensstemmelse med gældende krav og specifikationer for deres produkter. Kravene til disse kvalitetssystemer kan findes i kvalitetssystemet (QS)-forordningen i 21 CFR Part 820, hvis enheden sælges i USA, eller andre regler for andre lande (f.eks. EU MDR 2017/745 for EU).
Afhængigt af enhedens beskaffenhed kan QS-kravene være relevante i førmarkedsstadiet, postmarkedsstadiet eller begge dele. I forbindelse med præmarket-fasen kan demonstration af rimelig sikkerhed for sikkerhed og effektivitet for visse enheder med cybersikkerhedsrisici indebære at inkludere dokumentationsoutput relateret til QS-forordningen som en del af præmarket-indsendelsen. For eksempel pålægger ISO 13485:2016 og 21 CFR 820, at producenter af alle klasser af enheder, der er automatiseret med software, etablerer procedurer til at kontrollere designet af enheden, der sikrer overholdelse af specificerede designkrav (benævnt "designkontroller"). Inden for designkontroller er producenterne forpligtet til at "etablere og vedligeholde procedurer til validering af enhedsdesignet", som omfatter "softwarevalidering og risikoanalyse, hvor det er relevant". Som en del af den påbudte softwarevalidering og risikoanalyse kan softwareenhedsproducenter være nødt til at indføre cybersikkerhedsrisikostyring og -valideringsprocesser, når det er relevant.
Softwarevalidering og risikostyring udgør afgørende elementer i cybersikkerhedsanalyser, der afgør, om en enhed giver rimelig sikkerhed for sikkerhed og effektivitet. FDA giver producenterne mandat til at inkorporere udviklingsprocesser, der tager højde for og adresserer softwarerisici i hele design- og udviklingsstadierne som en del af designkontrol. Disse processer bør omfatte cybersikkerhedsovervejelser. Dette omfatter adressering af identifikation af sikkerhedsrisici, etablering af designkrav til styring af disse risici og fremskaffelse af bevis for, at kontrollerne fungerer efter hensigten og er effektive i enhedens udpegede miljø, hvilket sikrer tilstrækkelige sikkerhedsforanstaltninger.
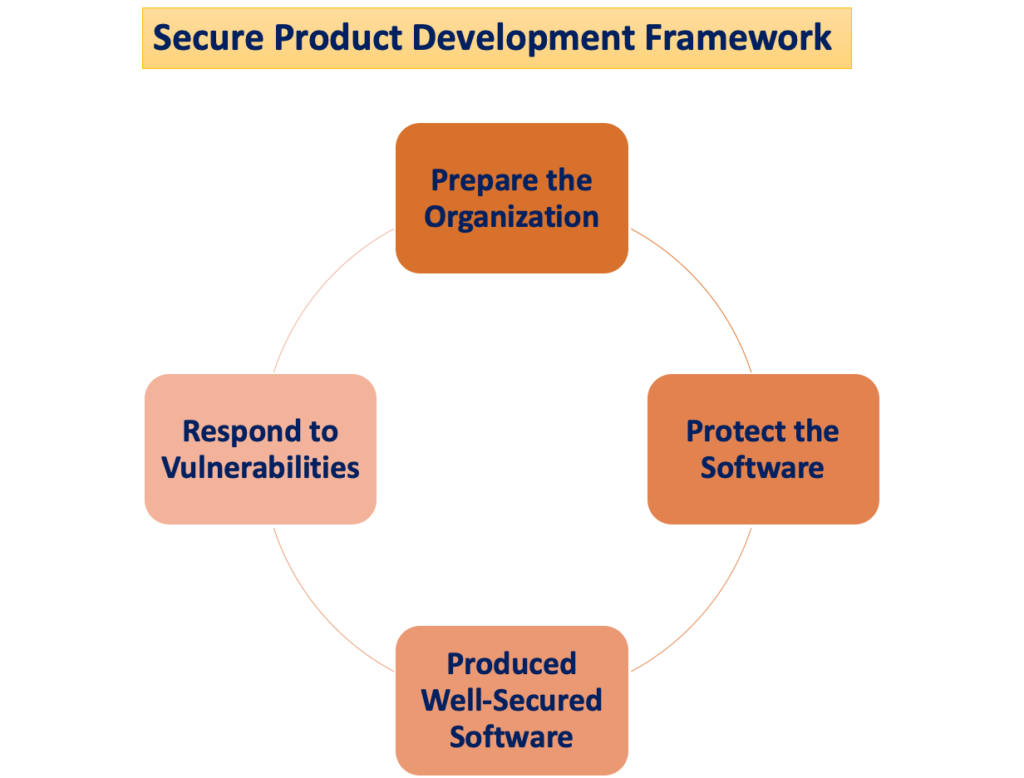
Den "Sikker produktudviklingsramme"
Potentialet for patientskade opstår, når cybersikkerhedstrusler udnytter sårbarheder i et system, og den lethed, hvormed disse trusler kan kompromittere sikkerheden og effektiviteten af et medicinsk udstyr, stiger med antallet af identificerede sårbarheder over tid. A Secure Product Development Framework (SPDF) består af processer, der har til formål at identificere og mindske mængden og alvoren af sårbarheder i produkter. Omfatter alle stadier af et produkts livscyklus – design, udvikling, frigivelse, support og nedlukning – SPDF er integreret.
Under enhedsdesign kan inkorporering af SPDF-processer forhindre nødvendigheden af re-engineering, når der integreres post-marketing-forbindelsesbaserede funktioner eller adressering af sårbarheder, der udgør ukontrollerede risici. Gennemførlig integration med eksisterende processer til produkt- og softwareudvikling, risikostyring og det bredere kvalitetssystem bidrager til SPDF'ens alsidighed.
For at sikre overholdelse af kvalitetssystemet (QS)-forordningen anbefales brugen af en SPDF. FDA opfordrer producenterne til at omfavne SPDF for dens fordele ved at opfylde både QS-forordningen og cybersikkerhedskravene. Det anerkendes dog, at alternative tilgange også kan opfylde QS-forordningen.
Håndtering af cybersikkerhedsrisici
Det primære formål med at anvende en SPDF (Secure Product Development Framework) er at skabe og vedligeholde enheder, der er både sikre og effektive. Fra et sikkerhedsperspektiv opnår disse enheder også pålidelighed og modstandsdygtighed. Producenter og/eller brugere (f.eks. patienter, sundhedsfaciliteter) kan derefter administrere disse enheder, herunder installation, konfiguration, opdateringer og gennemgang af enhedslogfiler, gennem enhedsdesignet og tilhørende mærkning.
Sundhedsfaciliteter har mulighed for at administrere disse enheder inden for deres egne cybersikkerhedsrisikostyringsrammer, såsom det bredt anerkendte National Institute of Standards and Technology (NIST) Framework for Improving Critical Infrastructure Cybersecurity, almindeligvis omtalt som NIST Cybersecurity Framework eller NIST CSF.
FDA anbefaler, at producenterne inkorporerer enhedsdesignprocesser, såsom dem, der er beskrevet i Quality System (QS)-forordningen, for at styrke sikker produktudvikling og vedligeholdelse. Mens de opretholder fleksibiliteten for producenterne, kan de også udforske alternative rammer, der stemmer overens med QS-forordningen og overholder FDA-anbefalinger for implementering af en SPDF. Eksempler inkluderer den medicinske udstyrsspecifikke ramme i Medical Device and Health IT Joint Security Plan (JSP) 30 og IEC 81001-5-1. Rammer fra andre sektorer, såsom ANSI/ISA 62443-4-1 Sikkerhed for industriel automation og kontrolsystemer Del 4-1: Produktsikkerhedsudvikling livscykluskrav, kan også opfylde QS-regler.
I de følgende afsnit af denne artikel gives anbefalinger til brug af SPDF-processer, som generelt opfattes af FDA, som fremhæver afgørende overvejelser for at udvikle enheder, der er sikre og effektive. Disse processer supplerer QS-forordningen, og FDA foreslår, at producenterne inkluderer tilsvarende dokumentation til gennemgang i præmarket-indsendelser.
Cybersikkerhedstrusselmodel
Trusselsmodellering involverer en systematisk proces til at identificere sikkerhedsmål, risici og sårbarheder i hele det medicinske udstyrssystem. Efterfølgende indebærer det, at der defineres modforanstaltninger for at forhindre, afbøde, overvåge eller reagere på virkningerne af trusler gennem hele det medicinske udstyrs systems livscyklus. Når den anvendes korrekt og omfattende, tjener den som grundlaget for at optimere sikkerheden på tværs af systemkomponenter, produkter, netværk, applikationer og forbindelser.
Med hensyn til styring af sikkerhedsrisici og for at lokalisere passende sikkerhedsrisici og kontroller for det medicinske udstyrssystem, anbefaler FDA implementeringen af trusselsmodellering for at informere og understøtte risikoanalyseaktiviteter. I forbindelse med risikovurdering foreslår FDA at integrere trusselsmodellering gennem hele designprocessen, der omfatter alle elementer i det medicinske udstyrssystem.
Nøgleaspekter af trusselsmodellen bør omfatte identifikation af risici og afbødninger og informere både før- og efterafbødningsrisici, der tages i betragtning i vurderingen af cybersikkerhedsrisikoen. Derudover bør modellen formulere antagelser om det medicinske udstyrssystem eller brugsmiljøet, såsom at antage, at hospitalsnetværk i sagens natur er fjendtlige. Denne antagelse får producenterne til at overveje scenarier, hvor en modstander kontrollerer netværket og er i stand til at ændre, droppe og afspille pakker. Desuden bør trusselsmodellen fange cybersikkerhedsrisici introduceret gennem forsyningskæden, fremstilling, udrulning, interoperation med andre enheder, vedligeholdelses-/opdateringsaktiviteter og nedlukningsaktiviteter, som kan blive overset i en traditionel sikkerhedsrisikovurderingsproces.
Til præmarket-indsendelser anbefaler FDA at inkludere trusselsmodelleringsdokumentation for at fremvise analysen af det medicinske udstyrssystem og identificere potentielle sikkerhedsrisici, der kan påvirke sikkerhed og effektivitet. Producenterne har fleksibiliteten til at vælge mellem forskellige metoder eller kombinationer af metoder til trusselsmodellering, og begrundelsen for deres valgte metoder bør forsynes med dokumentationen.
Det anbefales at udføre trusselsmodelleringsaktiviteter under designgennemgange, og dokumentationen bør give tilstrækkelig information til, at FDA kan vurdere og gennemgå sikkerhedsfunktionerne integreret i enheden. Denne holistiske evaluering bør tage hensyn til både enheden og det bredere system, den fungerer i, og understrege enhedens sikkerhed og effektivitet.
Hovedelementerne i cybersikkerhedstrusselmodellerne kan opsummeres som følger:
Identifikation af potentielle trusler
Udviklingen af en trusselsmodel tjener som et grundlæggende trin i cybersikkerhedsprocessen, der gør det muligt for producenter at identificere og forstå potentielle cybersikkerhedstrusler, der er specifikke for deres medicinske udstyr. Udviklingen af en trusselsmodel tjener som et grundlæggende trin i cybersikkerhedsprocessen, der gør det muligt for producenter at identificere og forstå potentielle cybersikkerhedstrusler, der er specifikke for deres medicinske udstyr. Udviklingen af en trusselsmodel tjener som et grundlæggende trin i cybersikkerhedsprocessen, der gør det muligt for producenter at identificere og forstå potentielle cybersikkerhedstrusler, der er specifikke for deres medicinske udstyr.
Risikovurdering og styring
Risikovurdering og -styring involverer evaluering af de identificerede trusler for at fastslå deres potentielle indvirkning og udformning af passende strategier til at afbøde disse risici effektivt.
Implementering af sikkerhedskontrol
Sikkerhedskontrol er de sikkerhedsforanstaltninger eller modforanstaltninger, der er implementeret for at beskytte det medicinske udstyrs fortrolighed, integritet og tilgængelighed mod identificerede trusler.
Fremtidige tendenser i FDA-retningslinjer for cybersikkerhed
Efterhånden som trusler og teknologi udvikler sig, vil FDA's retningslinjer for cybersikkerhed også udvikle sig. Det er vigtigt for producenterne at forblive informerede og agile over for disse ændringer. Producenter bør forudse opdateringer af reglerne ved at holde sig ajour med branchetendenser og opretholde en proaktiv tilgang til cybersikkerhed.
Forberedelse til uforudsete udfordringer er nøglen; etablering af robuste sikkerhedsprotokoller og fremadrettede strategier vil positionere producenterne til effektivt at reagere på den fremtidige tilstand af cybersikkerhedsregler.
Cybersikkerhed er et aspekt af regulering af medicinsk udstyr, som ikke kan overvurderes – det er lige så iboende for enhedssikkerhed som enhver mekanisk eller elektrisk funktion. FDA har erkendt dette, og ved at implementere en omfattende cybersikkerhedsramme sigter den mod at holde trit med innovation og samtidig beskytte folkesundheden. Producenter, sundhedspersonale og patienter skal alle samarbejde for at opretholde disse standarder og sikre den fortsatte pålidelighed og sikkerhed af medicinsk udstyr i forhold til cybertrusler.
Abonner på QualityMedDev nyhedsbrev
QualityMedDev er en online platform med fokus på kvalitet og reguleringsmæssige emner for virksomhed med medicinsk udstyr; Følg os på LinkedIn , Twitter for at holde dig ajour med de vigtigste nyheder om reguleringsområdet.
QualityMedDev er en af de største online platforme, der understøtter forretninger inden for medicinsk udstyr til emner vedrørende overholdelse af lovgivning. Vi sørger for reguleringsrådgivning over en bred vifte af emner, fra EU MDR & IVDR til ISO 13485, herunder risikostyring, biokompatibilitet, brugervenlighed og softwareverifikation og -validering og generelt support ved udarbejdelse af teknisk dokumentation til MDR.
Vores søsterplatform QualityMedDev Academy giver mulighed for at følge online- og træningskurser i eget tempo, der fokuserer på lovoverholdelses-emner for medicinsk udstyr. Disse uddannelseskurser, udviklet i samarbejde med højt kvalificerede fagfolk i sektoren for medicinsk udstyr, giver dig mulighed for eksponentielt at øge dine kompetencer over en bred vifte af kvalitets- og regulatoriske emner til forretningsdrift inden for medicinsk udstyr.

Tøv ikke med at tilmelde dig vores nyhedsbrev!
- SEO Powered Content & PR Distribution. Bliv forstærket i dag.
- PlatoData.Network Vertical Generative Ai. Styrk dig selv. Adgang her.
- PlatoAiStream. Web3 intelligens. Viden forstærket. Adgang her.
- PlatoESG. Kulstof, CleanTech, Energi, Miljø, Solenergi, Affaldshåndtering. Adgang her.
- PlatoHealth. Bioteknologiske og kliniske forsøgs intelligens. Adgang her.
- Kilde: https://www.qualitymeddev.com/2024/01/26/cybersecurity-threat-model/
- :har
- :er
- :ikke
- :hvor
- $OP
- 2016
- 30
- 350
- 820
- 9
- a
- Om
- Academy
- erkendte
- tværs
- aktiviteter
- Derudover
- adresse
- adressering
- Tilføjer
- klæbe
- administration
- rådgivet
- fortalere
- adræt
- Rettet
- målsætninger
- tilpasse
- Alle
- tillader
- også
- alternativ
- an
- analyser
- analyse
- ,
- foregribe
- enhver
- anvendelig
- applikationer
- anvendt
- Indløs
- tilgang
- tilgange
- passende
- passende
- ER
- artikel
- AS
- udseende
- aspekter
- vurdere
- vurdering
- forbundet
- antagelse
- antagelser
- sikkerhed
- At
- Angreb
- Automatiseret
- Automation
- tilgængelighed
- BE
- bliver
- blive
- været
- fordele
- styrke
- både
- bred
- bredere
- bragte
- virksomhed
- forretningsdrift
- men
- by
- CAN
- kan ikke
- stand
- fange
- vis
- kæde
- udfordringer
- Ændringer
- Vælg
- valgt
- klasser
- samarbejde
- samarbejde
- KOM
- kombinationer
- almindeligt
- komplement
- Compliance
- komponenter
- omfattende
- kompromis
- design
- Bekymringer
- fortrolighed
- Konfiguration
- tilsluttet
- tilsluttede enheder
- Tilslutninger
- Overvej
- overvejelser
- betragtes
- konsekvent
- består
- udgøre
- rådgivning
- moderne
- sammenhæng
- fortsatte
- kontrol
- styring
- kontrol
- Tilsvarende
- kunne
- lande
- kurser
- dækker
- skabe
- kritisk
- Kritisk infrastruktur
- afgørende
- Cyber
- Cyberangreb
- Cybersecurity
- Dato
- definere
- definition
- demonstrerer
- implementering
- Design
- designproces
- udpeget
- bestemmelse
- udviklet
- udvikling
- Udvikling
- enhed
- Enheder
- forskellige
- digital
- faldende
- diskuterer
- dokumentation
- Dropper
- medicin
- i løbet af
- e
- tidligste
- tjene
- lette
- Effektiv
- effektivt
- effektivitet
- enten
- elementer
- omfavne
- at understrege
- anvendelse
- muliggør
- Encompass
- vedrører generelt
- omfatter
- tilskynder
- sikre
- sikring
- Miljø
- væsentlig
- etablere
- etableret
- oprettelse
- Ether (ETH)
- EU
- europæisk
- europæiske Union
- evaluere
- evaluering
- NOGENSINDE
- bevismateriale
- udvikle sig
- eksempler
- eksisterende
- Exploit
- udforske
- eksponentielt
- Ansigtet
- faciliteter
- fda
- gennemførlig
- Feature
- Funktionalitet
- felt
- Fleksibilitet
- fokuserede
- følger
- efter
- følger
- mad
- Food and Drug Administration
- Food and Drug Administration (FDA)
- Til
- forkant
- fremadrettet
- fundet
- Foundation
- foundational
- Framework
- rammer
- fra
- fuld
- funktionalitet
- Endvidere
- fremtiden
- Generelt
- generelt
- retningslinjer
- skade
- Have
- Helse
- sundhedspleje
- Høj
- Fremhæv
- stærkt
- holistisk
- Hospital
- Men
- HTML
- HTTPS
- Identifikation
- identificeret
- identificere
- identificere
- if
- KIMOs Succeshistorier
- Påvirkninger
- implementering
- implementeret
- gennemføre
- vigtigt
- forbedring
- in
- omfatter
- omfatter
- Herunder
- indarbejde
- inkorporering
- Forøg
- Stigninger
- stigende
- industrielle
- industriel automation
- industrien
- informere
- oplysninger
- informeret
- Infrastruktur
- sagens natur
- initial
- Innovation
- installation
- instans
- Institut
- integral
- integreret
- Integration
- integration
- integritet
- beregnet
- ind
- iboende
- introduceret
- involvere
- involverer
- ISO
- IT
- ITS
- fælles
- Holde
- Nøgle
- mærkning
- største
- livscyklus
- livscyklus
- MailChimp
- Main
- vedligeholde
- opretholdelse
- vedligeholdelse
- administrere
- ledelse
- mandater
- Producenter
- Produktion
- max-bredde
- Kan..
- MDR
- foranstaltninger
- mekanisk
- medicinsk
- medicinsk udstyr
- medicinsk udstyr
- Mød
- møde
- metoder
- metoder
- måske
- afbøde
- formildende
- afbødende risici
- model
- modellering
- modeller
- Overvåg
- mere
- mest
- skal
- national
- Natur
- Navigation
- nødvendighed
- Behov
- netværk
- net
- nyheder
- NIST
- nummer
- objektiv
- målsætninger
- of
- on
- ONE
- online
- kun
- betjene
- opererer
- Produktion
- optimering
- Option
- or
- Andet
- vores
- skitseret
- udgange
- i løbet af
- Tilsyn
- egen
- Tempo
- pakker
- del
- patient
- patienter
- opfattet
- perspektiv
- fase
- afgørende
- fly
- perron
- plato
- Platon Data Intelligence
- PlatoData
- spiller
- plugin
- stillet
- position
- Muligheden
- Indlæg
- potentiale
- praksis
- forberedelse
- forhindre
- primære
- Proaktiv
- procedurer
- behandle
- Processer
- Produkt
- produktudvikling
- Produkter
- professionelle partnere
- prompter
- beskytte
- beskyttelse
- protokoller
- give
- forudsat
- giver
- leverer
- offentlige
- folkesundheden
- offentliggjort
- kvalitet
- mængde
- rækkevidde
- rationale
- rimelige
- anerkendt
- anbefalinger
- anbefales
- anbefaler
- benævnt
- Regulering
- regler
- lovgivningsmæssige
- Regulatory Compliance
- relaterede
- relation
- frigive
- pålidelighed
- forblive
- påkrævet
- Krav
- modstandskraft
- Svar
- gennemgå
- Anmeldelser
- Rise
- Risiko
- risikovurdering
- risikostyring
- risici
- robust
- roller
- sikker
- varetagelse
- sikkerhedsforanstaltninger
- Sikkerhed
- samme
- scenarier
- sektioner
- sektor
- Sektorer
- sikker
- sikkerhed
- Sikkerhedsforanstaltninger
- styring af sikkerhedsrisiko
- sikkerhedsrisici
- tjener
- flere
- sværhedsgrad
- bør
- udstillingsvindue
- søster
- faglært
- smartere
- So
- Software
- softwareudvikling
- solgt
- specifikke
- specifikationer
- specificeret
- Stage
- etaper
- standarder
- Tilstand
- Stater
- forblive
- opholder
- Trin
- strategier
- indsendelse
- Bidrag
- Hold mig opdateret
- Efterfølgende
- sådan
- tilstrækkeligt
- foreslår
- egnede
- forsyne
- forsyningskæde
- support
- Støtte
- systemet
- Systemer
- Teknisk
- Teknologier
- end
- at
- Fremtiden
- deres
- derefter
- Disse
- de
- denne
- dem
- trussel
- trusler
- Gennem
- hele
- tid
- gange
- til
- også
- Emner
- traditionelle
- Kurser
- Tendenser
- Stol
- troværdighed
- forstå
- uforudset
- union
- Forenet
- Forenede Stater
- opdateringer
- Opretholde
- URL
- us
- usability
- Brug
- brug
- brugere
- Ved hjælp af
- validering
- validering
- forskellige
- Verifikation
- alsidighed
- Sårbarheder
- Sårbar
- we
- websites
- hvornår
- hvorvidt
- som
- mens
- bredt
- vilje
- med
- inden for
- WordPress
- WordPress plugin
- dig
- Din
- zephyrnet





